
Evolution des maladies inflammatoires : revue
dernière mise à jour le 12/02/2021
L'association entre inflammation et maladies de l'homme moderne (obésité, maladies cardiovasculaires, diabète de type 2, cancer) reste mystérieuse. L'inflammation est une réponse de protection contre des stimuli nocifs qui entraîne inévitablement un coût dans le fonctionnement tissulaire normal. Ce compromis fondamental entre le coût et les avantages de la réponse inflammatoire a été optimisé par l'évolution au fil du temps et des conditions environnementales spécifiques. L'évolution rapide de l’environnement humain due à la construction de niches qui débordent les capacités d’adaptation génétique par sélection naturelle, conduisent de plus en plus à une inadéquation entre l'environnement moderne
et les traits sélectionnés. Par conséquent, plusieurs compromis de la physiologie humaine ne sont plus optimisés pour l'environnement moderne, conduisant à une augmentation de la susceptibilité à certaines maladies.
Nous examinons ici la réponse inflammatoire dans une perspective évolutionniste. Nous discutons ici les aspects uniques de la réponse inflammatoire et son histoire évolutionniste qui peut nous aider à expliquer l'association entre l'inflammation et les maladies humaines modernes.
Introduction
L'inflammation est classiquement décrite comme une réponse à une infection ou à une blessure. Il est maintenant de plus en plus avéré que les maladies inflammatoires chroniques sont universellement associée aux maladies de la richesse et de la longue durée de vie telles que l'obésité [1], les maladies cardiovasculaires [2], les maladies neurodégénératives [3], et le cancer [4]. La prévalence de ces maladies a augmenté rapidement au cours des dernières décennies [5].
Ceci soulève deux questions importantes. Quels sont les aspects de la biologie humaine qui nous rendent sensibles à ces maladies et pourquoi ces maladies aux étiologies disparates sont-elles toutes associées à une inflammation chronique ? Les raisons d’une association intime entre inflammation et pathologie sont bien comprises dans les cas extrêmes de dérégulation de la réponse inflammatoire, comme constatés dans certains dommages tissulaires et dans les septicémies [6,7]. Dans ces conditions, le rôle normalement protecteur de l'inflammation devient néfaste par un excès d’intensité ou de durée. L’anaphylaxie et le choc septique sont les deux exemples les plus connus de réponse inflammatoire potentiellement mortelle. Cependant, bien que l'inflammation excessive puisse clairement être pathogène, l'association quasi-universelle entre inflammation et maladies humaines ne peut être expliquée seulement par une dérégulation. Les caractéristiques fondamentales des processus inflammatoires qui contribuent à la susceptibilité aux maladies sont encore mal compris, comme le sont les caractéristiques des tissus et organes cibles les plus vulnérables aux pathologies inflammatoires.
Nous discutons ici de l’inflammation et des maladies inflammatoires du point de vue évolutionniste. Nous nous concentrons sur le compromis coût-bénéfice de la réponse inflammatoire et discutons de l’impact des changements environnementaux sur la susceptibilité aux maladies humaines. Bref nous abordons l’inflammation sous l’aspect des sciences de l’évolution.
Inflammation et maladies
Une réponse inflammatoire peut être déclenchée par une variété de stimuli nocifs, comme l'infection et les blessures. En conséquence, les réponses inflammatoires sont très hétérogènes
en termes de types cellulaires et de médiateurs moléculaires impliqués. L'inflammation se présente aussi sous différents modes : aiguë ou chronique, locale ou systémique [8]. Malgré cette complexité, toutes les réponses inflammatoires peuvent être décomposées en quatre entités qui constituent la voie inflammatoire universelle : inducteurs, capteurs, médiateurs, et cible tissulaire [9].
Les inducteurs peuvent être exogènes (agents pathogènes ou toxines [10]) ou endogènes (ATP ou cristaux d'urate [11]), qui provoquent stress, blessures ou dysfonctionnement cellulaire [12]. Les capteurs cellulaires sont les macrophages et mastocytes, présents dans tous les tissus, dont certains récepteurs spécifiques détectent les inducteurs et qui répondent en produisant des médiateurs inflammatoires. En fonction de la nature des inducteurs, les cellules de capteurs produisent différentes combinaisons et quantités de médiateurs, créant une signature unique de médiateur pour chaque inducteur. Les médiateurs de l’inflammation agissent, à leur tour, sur les tissus cibles et modifient leurs états fonctionnels, en promouvant l'élimination des inducteurs, l'adaptation à leur nocivité, et la restauration de l’homéostasie tissulaire [9].
L'altération des états fonctionnels des tissus cibles se produit généralement aux dépens de la fonction des tissus normaux [12-16], représentant ainsi le cinquième signe cardinal de l'inflammation
‘Functio laesa’ (perturbation de la fonction) [17]. Cette propriété fondamentale des médiateurs explique le potentiel pathogène de la réponse inflammatoire. Les exemples d'altération fonctionnelles induites par l’inflammation sont : augmentation de l'adhésivité endothéliale
et à la perméabilité par la cytokine facteur de nécrose tumorale α (TNFα) et l'interleukine-1
b (IL-1b), pour permettre la formation d'exsudat [6]; augmentation de production de mucus par les cellules caliciformes, en réponse à l'IL-13, pour améliorer les défenses de la barrière épithéliale [18]; et synthèse et sécrétion par les hépatocytes de protéines de phase aiguë (PPA tels que CRP, ferritine, fibrinogène, etc.) en réponse à l'IL-6, pour promouvoir défense de l'hôte contre l'infection [19]. Chaque de ces effets fournit un avantage adaptatif de réponse à l'infection; cependant, ils imposent un coût lié à la diminution de la fonction normale des tissus cibles. Dans les exemples cités, l’augmentation de la perméabilité endothéliale et de l’adhésivité peut nuire à l'hémostase [20], l'augmentation de la production de mucus peut conduire à la formation de bouchons muqueux qui réduisent les échanges gazeux par l’épithélium respiratoire [21,22], et l’augmentation de synthèse des APP conduit à une réduction de la synthèse de l'albumine et d'autres protéines sériques [19,23-25]. C’est l'altération des états fonctionnels des tissus et organes qui sous-tend le potentiel pathologique de la réponse inflammatoire. Comme ces modifications fonctionnelles sont une conséquence fondamentale de toute réponse inflammatoire, le coût et le potentiel pathologique existent indépendamment de l'ampleur et de la durée de la réponse inflammatoire. Ainsi, alors que les dommages tissulaires induits par une réponse inflammatoire excessive sont les plus notables des résultats négatifs, ils ne sont pas les seuls, ni même les plus courants dans le coût de cette réponse.
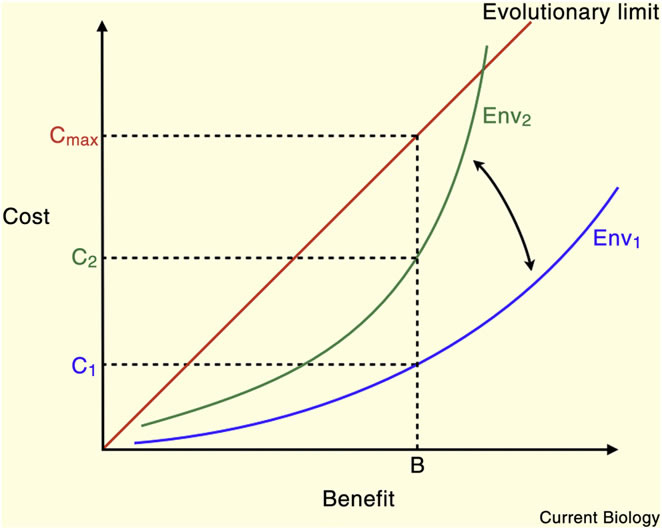 Figure 1. Les coûts et avantages des compromis sont optimisés pour des environnements spécifiques et peuvent être affectés par des changements environnementaux. Une caractéristique particulière peut être choisie et maintenue par une évolution aussi longtemps que le bénéfice du trait emporte sur le coût. Ici, le caractère existe dans Environnement 1 (Env1) avec un bénéfice B et un coût C1. Si l’environnement change, le coût de maintien du bénéfice B peut augmenter à C2. Dans toutes les conditions, la ligne rouge délimite une zone où les coûts sont inférieurs ou égaux aux bénéfices. Les traits situés en dessous et à droite de la ligne sont considérés comme adaptative et les traits au-dessus et à gauche sont inadaptés. Il existe cependant une limite supérieure pour un coût acceptable, c’est lorsque le trait devient préjudiciable indépendamment de son avantage. Ces traits sont définis par un coût absolu très élevé.
Figure 1. Les coûts et avantages des compromis sont optimisés pour des environnements spécifiques et peuvent être affectés par des changements environnementaux. Une caractéristique particulière peut être choisie et maintenue par une évolution aussi longtemps que le bénéfice du trait emporte sur le coût. Ici, le caractère existe dans Environnement 1 (Env1) avec un bénéfice B et un coût C1. Si l’environnement change, le coût de maintien du bénéfice B peut augmenter à C2. Dans toutes les conditions, la ligne rouge délimite une zone où les coûts sont inférieurs ou égaux aux bénéfices. Les traits situés en dessous et à droite de la ligne sont considérés comme adaptative et les traits au-dessus et à gauche sont inadaptés. Il existe cependant une limite supérieure pour un coût acceptable, c’est lorsque le trait devient préjudiciable indépendamment de son avantage. Ces traits sont définis par un coût absolu très élevé.
Les états fonctionnels des tissus et organes sont maintenus par une variété de mécanismes de contrôle homéostatique sous la dépendance du système endocrinien et du système nerveux [21,26]. Les hormones et neurotransmetteurs régulent les activités des tissus et organes pour maintenir des variables physiologiques clés, telles que la tension artérielle, le niveau de glucose ou la température corporelle, dans une certaine plage de valeurs souhaitées. Ces mécanismes de contrôle homéostatique permettent également l'adaptation à un environnement changeant ou à des besoins physiologiques particuliers [27]. Par exemple, le système nerveux autonome commande la thermogenèse et la dissipation de chaleur pour s’adapter à la température ambiante, il contrôle le débit sanguin pour l’orienter vers les muscles ou l’intestin selon les priorités courantes (chasse, fuite ou digestion) [28]. Les hormones pancréatiques, insuline et glucagon, contrôlent respectivement la néoglucogenèse et la glycogénolyse pour réguler la glycémie niveau en réponse au jeûne et à l'alimentation [29]. Les mécanismes homéostatiques contrôlent et maintiennent la fonction des tissus et organes en réponse à des changements environnementaux stéréotypés et à des ensembles prédéfinis de priorités physiologiques caractéristiques d’une espèce donnée dans une niche écologique donnée.
Toutefois, lorsque l'homéostasie est perturbée par des conditions nocives comme l'infection ou des lésions tissulaires, les mécanismes de contrôle homéostatiques deviennent insuffisants pour maintenir ou restaurer l'homéostasie. C’est dans ces conditions que les mécanismes inflammatoires de contrôle sont engagés afin d'éliminer le stimulus nociceptifs et de rétablir l'homéostasie. Afin de remplir ces fonctions, l’inflammation modifie les états fonctionnels des tissus et organes d'une manière qui est généralement opposée aux mécanismes de contrôle homéostatique. La raison de cet antagonisme tient à la différence des objectifs proximaux de l’homéostatique et de l’inflammation ; la première vise à maintenir des fonctions normales
tandis que la seconde vise à modifier temporairement les fonctions afin de faire face à un nouveau besoin ou à des conditions nocives. En plus d'être antagonistes, les mécanismes de l’inflammation dominent ceux de l’homéostasie [30]. La raison de cette domination des mécanismes de contrôle de l’inflammation sur ceux du contrôle de l’homéostasie est la plus grande priorité des objectifs de la première qui répond à des conditions qui menacent l'aptitude d'un organisme, voire sa survie. Par exemple, les médiateurs inflammatoires peuvent
induire des changements dans la température du corps, l'appétit, le sommeil, les seuils nociceptifs, le tonus des muscles lisses des voies vasculaires et respiratoires, et une variété de processus métaboliques [21]. Ces effets de médiateurs inflammatoires sont antagonistes
des contrôles homéostatiques induits par l'hypothalamus, le système nerveux autonome et les
hormones. Les changements des paramètres de l'homéostasie sont bénéfiques, car ils aident la défense contre les agressions, et coûteux, car ils interfèrent avec les fonctions normales quelle que soit l'intensité ou la durée de la réponse inflammatoire.
Ceci représente bien le potentiel pathologique de la réponse inflammatoire et souligne le compromis fondamental coût-bénéfice de l'inflammation. Ce que nous allons aborder plus en détail.
Compromis coûts-bénéfices de la réponse inflammatoire
Le compromis entre les aspects bénéfiques et néfastes de la réponse inflammatoire a plusieurs caractéristiques qui peuvent expliquer l'augmentation rapide de la prévalence des maladies inflammatoires dans les populations humaines modernes. Etant donné que ces caractéristiques
ne sont pas propres à l'inflammation, nous allons en discuter dans une perspective plus générale.
Le compromis coût-bénéfice d'un trait donné est optimisé pour maximiser les avantages tout en minimisant le coût. Cette optimisation est souvent spécifique pour un environnement particulier: le trait peut être optimal dans un environnement et sous-optimal ou même préjudiciable dans d'autres environnements. Ainsi, les modifications appropriées aux aspects les plus pertinents de l'environnement peuvent altérer l’équilibre du rapport coût-bénéfice, en le rendant sous-optimal et, à l'extrême, inadapté si le coût l'emporte sur les bénéfices. Si la modification environnementale est stable, le trait peut être soit optimisé pour le nouvel environnement par la sélection naturelle, ou tout à fait perdu si l’impact est négatif sur l’aptitude à la reproduction. Le degré de sensibilité aux facteurs environnementaux peut aussi varier en fonction du trait lui-même. Certains traits, dont les métabolismes de base et les processus de développement, sont relativement indépendants de l'environnement. D’autres peuvent être très sensibles aux changements environnementaux, ce sont ceux qui sont directement à l’interface avec l'environnement, comme les réponses immunitaires et inflammatoires. En effet, le compromis coût-bénéfice de réponse inflammatoire peut être affecté par de nombreux facteurs environnementaux comme une altération de l'exposition aux microorganismes commensaux et pathogènes due à la densité démographique, les changements alimentaires, l’hygiène, les antibiotiques et les vaccins. Comme beaucoup de ces facteurs ont changé radicalement au cours des derniers siècles, on peut s'attendre à ce que le compromis coût-bénéfice de la réponse inflammatoire dans les populations humaines modernes ne soit plus optimisé dans l'environnement actuel.
Généralement, le bénéfice d'un trait définit le coût acceptable : aussi longtemps que le bénéfice est supérieur au coût le trait peut être sélectionné et maintenu par l'évolution. Cela est vrai jusqu'à un certain point : le coût peut atteindre une limite physiologique qui affecte la survie ou la reproduction et il devient inacceptable indépendamment des bénéfices associés (Figure 1). Dans ces limites, les traits à haute prestation (survie ou reproduction) peuvent être associés à des coûts élevés. Bien que, dans un environnement donné les avantages d'un trait soient plus élevés que les coûts, les traits diffèrent en termes de valeur absolue des coûts et des avantages : il y a des traits à haut coût et hauts avantages et des traits à faible coût et faibles avantages. Parce qu’un changement environnemental peut modifier l'équilibre coût-bénéfice optimal, les traits de haute prestation à coûts et bénéfices tous deux élevés, devrait généralement être plus vulnérables aux changements environnementaux. En revanche, les traits qui ont un coût très faible peuvent être maintenus comme des atavismes, même lorsque les avantages offerts par le trait ont disparu. Des traits non adaptatifs peuvent également être maintenus par la sélection sexuelle ou par le biais de leur association avec différents traits bénéfiques. Par conséquent, tous les traits et leur compromis associé forment un spectre défini par leur sensibilité à l'évolution de tout facteur environnemental donné (figure 2). En conséquence, un changement donné dans l'environnement devrait affecter différents compromis à des degrés divers.
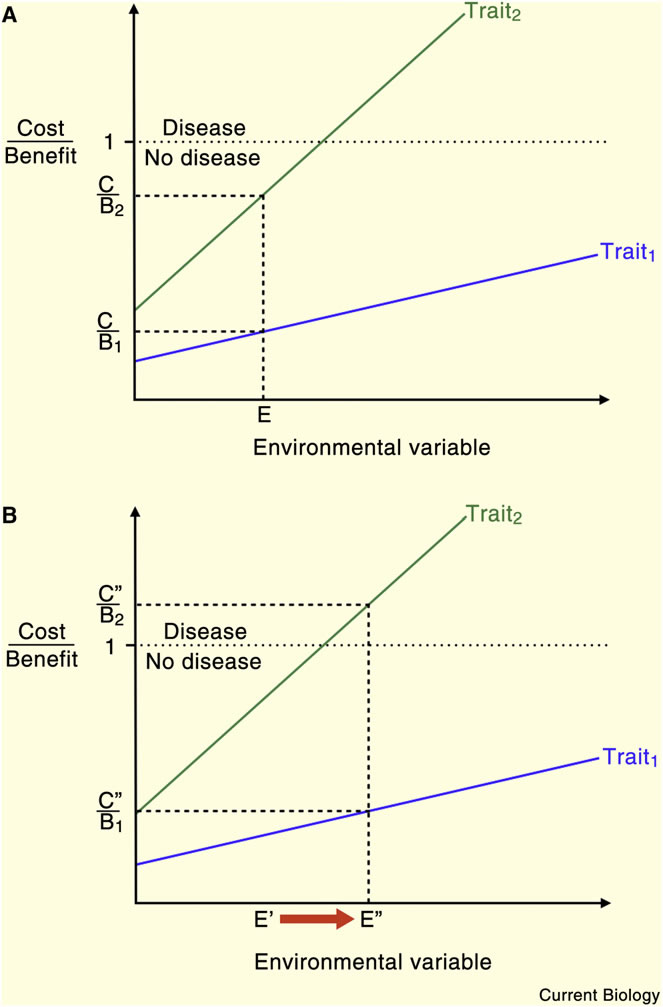 Figure 2. Les traits individuels sont caractérisés par des ratios coût-bénéfice (C / B qui doivent être 1).
Figure 2. Les traits individuels sont caractérisés par des ratios coût-bénéfice (C / B qui doivent être 1).
La réponse inflammatoire est un trait à haut coût et haut bénéfice : bénéfice élevé, car il peut sauver la vie face à des défis nocifs, coût élevé parce que les défenses inflammatoires interférent avec les fonctions normales et, à l'extrême, peuvent endommager les tissus et provoquer la mort. Par conséquent, la réponse inflammatoire est particulièrement vulnérable à des changements de facteurs environnementaux pertinents, comme par exemple des modifications d’exposition microbienne, d’alimentation, de stress, de toxines ou d’activité physique. Ceci, à son tour, contribue probablement à l’augmentation spectaculaire de la prévalence des maladies inflammatoires au cours des dernières décennies.
Coûts directs et vulnérabilités
En considérant l'impact de l'équilibre coût-bénéfice sur les maladies humaines, il peut être utile de faire la distinction entre deux types de coût: les coûts directs et les vulnérabilités. Les coûts directs sont inévitables, ce sont des coûts récurrents qui s’accumulent continuellement par déploiement ou maintenance d'un trait. Les vulnérabilités sont des conséquences rares mais catastrophiques d'un trait dans une fraction de la population. En termes de bilan, les coûts directs sont optimisés en réduisant leur valeur absolue tandis que les vulnérabilités sont optimisées en réduisant leur fréquence. Inversement, les changements environnementaux peuvent augmenter, soit la valeur des coûts directs, soit les risques de subir les conséquences des vulnérabilités. Cependant, la différence entre coûts directs et vulnérabilités n’est pas absolue : si les vulnérabilités deviennent courantes, elles peuvent se transformer en coûts directs. Pour illustrer les différences entre coûts directs et vulnérabilités, faisons l'analogie avec la possession d'une voiture. Certains coûts directs associés à l'avantage de posséder une voiture sont les frais d'essence, d'assurance, et de parking ; la vulnérabilité est le risque d’accident fatal. Les deux types de coûts peuvent augmenter ou diminuer en fonction de deux facteurs internes (efficacité du moteur et sécurité de la voiture) et de facteurs externes (densité du trafic et des mauvais conducteurs). En outre, les coûts directs et les vulnérabilités peuvent atteindre un point où ils l'emportent sur l'avantage de posséder une voiture. De même, les coûts directs et les vulnérabilités des traits biologiques peuvent contribuer à une susceptibilité accrue aux maladies humaines dans le cadre de compromis suboptimaux causés par des changements d'environnement. Dans la section suivante nous discutons de plusieurs exemples de traits humains et la susceptibilité aux maladies qui leur est associée.
Compromis anatomiques et physiologiques
Un éventail de maladies humaines communes reflète, en partie, le les coûts directs et les vulnérabilités de différents traits anatomie et physiologie de l'homme. Ici, nous discutons quatre exemples qui illustrent la façon dont les coûts et les vulnérabilités de traits spécifiques ont un impact direct sur la susceptibilité aux maladies inflammatoires. D'autres exemples de coûts directs et de vulnérabilités sont donnés dans le tableau 1 (ci-après).
| Trait | Bénéfice adaptatif | Coût direct | Vulnérabilité |
| Régénération épithéliale | Remplace constamment les tissus, produit des tissus neufs et fonctionnel, et répare les tissus | Coût métabolique d'une régénération continuelle [21] | Lente accumulation de mutations conduisant au développement de cancers [69] |
| Métabolisme aérobie | Augmentation considérable de production d'énergie [35] | Production de dérivés réactifs de l'oxygène [21] | Maladies neurodégénératives, sénescence cellulaire [5,21,70] |
| Antioxydants (ex : acide urique) |
Protège contre les dégâts des dérivés réactifs de l'oxygène [71] | Maintien d'un taux d'urate sérique optimal entre production et résorption rénale [72] | Prédispose à la goutte et à l'insuffisance rénale [37,38,41,42] |
| Endosquelette | Solide système de soutien. Supprime les contraintes de volume et de taille. Augmente les types de mouvements [31] | Absence de protection par exosquelette [31] | Embolie graisseuse, ostéoporose, fractures [5,21] |
| Articulations synoviales | Augmente l'ampleur et la liberté des mouvements [31] | L'usure peut conduire à l'arthrose [33] | Développement d'arthrite inflammatoire [33] |
| Hémostase | Protège contre les hémorragies induites par les traumatismes [21] | Augmente la viscosité du plasma, donc l'effort cardiaque [46,47,74] | L'activation inappropriée peut conduire à l'ischémie tissulaire (cerveau, cœur) |
| Inflammation | Restaure l'homéostasie. Elimine les agents pathogènes [12] | La production de dérivés réactifs de l'oxygène conduit à des pathologies immunitaires et des dommages tissulaires localisés [43] | L'hyperactvation peut provoquer un réponse systémique et un choc septique [45] |
| Immunité adaptative | Permet l'élimination des pathogènes sur leurs antigènes spécifique et conserve la mémoire des infections précédentes [8] | Maintenance d'une population de lymphocytes naïfs et mémoire [8] | Activation inappropriée contre ses propres antigènes qui peut conduire aux pathologie auto-immunes [8] |
| Microbiote | Amélioration de production d'énergie à partir de l'environnement. Production de micronutriments que l'organisme ne peut produire [75] | La barrière muqueuse et les leucocytes sont coûteux à produire [76] et peuvent gêner l'absorption de nutriments systémiques [77] | Colites inflammatoires [78] et infections récurrentes. |
| Epithéliums muqueux | Protection contre les particules et les infection [5,21] | Diminution des fonctions et de la diffusion [77] | Asthme et allergies [79] |
| Stockage d'énergie | Capacité de résistance au jeûne et à l'insécurité nutritionnelle [80] | Risque d'être une proie plus facile par limitation des mouvements [80] | L'absence de prédation peut conduire au DT2 et à l'obésité [80] |
Exosquelette et Endosquelette
L'exosquelette et l’endosquelette représentent deux différentes solutions dans le règne animal pour le support mécanique et la motilité. Le choix d'un type particulier de squelette détermine le plan et les fonctions du corps comme une contrainte générale sur d'autres changements évolutionnistes [31]. Les animaux à exosquelettes sont mieux protégés contre les lésions traumatiques, au prix d’une mobilité réduite, tandis que les animaux avec endosquelettes profitent d’une meilleure mobilité de différentes parties du corps, mais ils sont plus vulnérables aux blessures (à l'exception du cerveau où le crâne est assimilable à un exosquelette). En outre, les exosquelettes imposent d'autres contraintes
sur la croissance, la dissipation de chaleur, la sécrétion, et l'échange de gaz, qui sont absentes chez les espèces à endosquelettes [32]. La liberté de mouvement accrue chez les animaux avec un endosquelette est plus nette au niveau des articulations synoviales [21]. Ces articulations se distinguent par la présence d'une cavité synoviale cavité contenant un liquide de lubrification et des plaques de cartilage sur chacun des os de l’articulation pour absorber les chocs et réduire les frottements.
Bien que les articulations synoviales aient de nombreux avantages mécaniques, elles présentent également des coûts directs et des vulnérabilités. Un coût direct de ce système est que le stress persistant sur le cartilage conduit à son érosion progressive et conduit à une friction directe d’os à os avec des lésions osseuses. Finalement, le dommage persistant conduit à une limitation des mouvements et à des douleurs [21,33]. Le coût direct des articulations synoviales est l'arthrose. L’espace confiné intra-articulaire le rend sensible à la formation d’exsudats inflammatoires. A contrario, les tissus mous peuvent résister à un gonflement causé par un exsudat. En outre, le recrutement de leucocytes dans l'articulation,
particulièrement de macrophages et de neutrophiles, peuvent créer un cycle inflammatoire autonome qui mène à un dysfonctionnement progressif de l'articulation et à sa destruction [6,7]. Cette situation est celle de la polyarthrite rhumatoïde qui représente une vulnérabilité de la synoviale des articulations.
Métabolisme oxydatif
L’augmentation de la concentration de l'oxygène atmosphérique a conduit l'évolution au métabolisme oxydatif, augmentant la production d'énergie efficacité et permettant l'évolution vers des corps de plus grande taille [34-36]. Un coût direct corollaire au métabolisme oxydatif est la production élevée de dérivés réactifs de l'oxygène, comme les radicaux libres, qui peuvent endommager les tissus [21]. Les dommages tissulaires induits par ces dérivés a conduit l'évolution à produire des antioxydants intracellulaire et extracellulaires qui aident à baisser le niveau de ces dérivés et à maintenir l'homéostasie tissulaire. Les primates ont des mécanismes antioxydants extracellulaires uniques, utilisant l’urate et la bilirubine comme principaux antioxydants plasmatiques [37,38]. Le niveau élevé d'urate chez les primates résulte d'une forme inactive de l'enzyme uricase qui convertit l’urate en allantoïne. Par conséquent, les niveaux d'acide urique plasmatique sont 10 fois plus élevés chez les primates que chez d'autres animaux, jusqu’à atteindre la limite de solubilité à la température corporelle [39]. Fait intéressant, la présence d'uricase inactive est spécifique des primates et associée à la perte de la capacité à synthétiser un autre antioxydant puissant, l'acide ascorbique [40], les obligeant à s’en procurer par d’autres moyens.
La présence de taux plasmatiques élevés d'urate expose les primates à des vulnérabilités uniques. Les niveaux d'urate sérique sont si élevés qu’ils sont à la limite de la précipitation en cristaux, surtout aux endroits où la température est légèrement inférieure à la température centrale du corps (par exemple les articulations des orteils). Ces cristaux d'urate précipités peuvent activer le complexe NALP3, inflammasome produisant l'IL-1ß et conduisant à la crise de goutte [41]. En outre, l'urate peut précipiter dans le rein, ce qui conduit à une insuffisance rénale aiguë potentiellement mortelle [42]. Dans l’ensemble, les hauts niveaux d'urate fonctionnent comme de puissants antioxydants, mais créent des vulnérabilités aux maladies. La propension à la cristallisation de l'urate est fortement
influencée par l'alimentation [5] et donc cette vulnérabilité devrait être très sensible aux changements environnementaux tels que la composition des aliments.
Réponse immunitaire
Parce que les infections microbiennes peuvent être mortelles, les défenses immunitaire ont un bénéfice élevé est les coûts acceptables peuvent être également élevés, comme nous l’avons vu avant. Les coûts directs de la réponse immunitaire comprennent son coût énergétique ainsi que des lésions tissulaires collatérales. Par exemple, la défense anti-bactérienne médiée par les neutrophiles implique la génération de très puissants dérivés réactifs de l’oxygène bactéricides comme l'hypochlorite, le superoxyde et le peroxyde d'hydrogène [43]. La génération de ces dérivés provoque des lésions tissulaires [6]. C’est une conséquence inévitable de la clairance bactérienne, qui est partiellement atténué par des mécanismes de protection et de réparation tissulaire [44]. En plus des coûts directs, cette réponse immunitaire rapide et efficace crée une vulnérabilité se manifestant par une hyperactivation immunitaire systémique. Cette vulnérabilité, connue sous le choc septique, conduit à une cascade de défaillances d'organes et de tissus en raison d'une mauvaise perfusion, d’une coagulation intravasculaire disséminée, et finalement d’un collapsus vasculaire [45]. Les maladies auto-immunes sont un exemple de la vulnérabilité du système immunitaire adaptatif, elles se produisent lorsqu'une réponse immunitaire est dirigée contre les propres antigènes d'un organisme [8].
La coagulation du sang
La cascade de coagulation est un autre trait associé à des coûts directs et des vulnérabilités. Un système circulatoire fermé présente l'avantage d’une distribution performante de nutriments et d’oxygène Cependant, cela crée un problème secondaire de maintien de l'hémostase pour assurer une fonction vasculaire optimale et pour favoriser la réparation d’un dommage vasculaire. Les composants du système de coagulation sont constitutivement présents dans le sang afin d'assurer une réponse hémostatique rapide en cas de lésion vasculaire. Par conséquent, un coût direct du système de coagulation est une augmentation de la viscosité du plasma qui, à son tour, augmente l’effort cardiaque et nécessite un investissement accru de ressources pour la taille et la fonction du coeur [46,47]. Le fibrinogène est une composante clé de la cascade de coagulation et sa concentration a été liée à la viscosité du plasma et à des maladies [48]. Les vulnérabilités du système de coagulation sont la thrombose, l'embolie, l'ischémie et l’AVC, qui peuvent être causée par une activation inappropriée de la cascade de coagulation [21].
Compromis d’histoire de vie
Les traits d'histoire de vie sont des caractéristiques qui affectent directement le succès reproductif en incluant l'âge et la taille de la maturité sexuelle, la durée de la période reproductive, le vieillissement, ainsi que le nombre et la taille de la progéniture [49]. L’allocation de ressources à l’une quelconque de ces fonctions est un compromis de valeur sélective et la stratégie d'allocation est spécifique à chaque interaction organisme-environnement. La théorie des traits d'histoire de vie a montré que l’équilibre des investissements en ressources pour ces traits a été façonné par la sélection naturelle pour maximiser le succès de la reproduction d'un organisme [49].
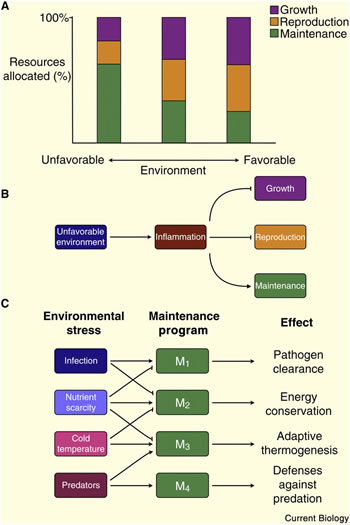 Figure 3. Le compromis entre les caractéristiques de l'histoire de la vie est une fonction de l'environnement.
Figure 3. Le compromis entre les caractéristiques de l'histoire de la vie est une fonction de l'environnement.
(A) Un environnement favorable favorise l'investissement des ressources dans la croissance et la reproduction et environnement défavorable favorise le maintien somatique au détriment de la croissance et la reproduction.
(B) Certains aspects d’un environnement défavorable, comme une infection ou une blessure, conduisent à une réponse inflammatoire qui à son tour favorise le maintien somatique et inhibe les voies qui contrôlent la croissance et la reproduction.
(C) Les différents stress environnementaux activent différente programmes de maintenance pour faire face à ce stress et ils inhibent les programmes incompatibles ou ceux qui sont liés par un compromis fonctionnel ou énergique.
L'investissement de ressources dans différents traits d'histoire de vie dépend de l'état de l'environnement. En général, un environnement favorable (par exemple, nourriture abondante, absence de prédateurs, température optimale) encourage l'investissement dans la croissance et / ou la reproduction, alors qu’un environnement défavorable favorise l'investissement dans les processus de réparation et de maintenance (figure 3A). Dans les cas extrêmes, les programmes d'entretien peuvent prendre des formes de vie suspendue, comme l'hibernation, l’estivation ou la diapause. Les traits d'histoire de vie d’Homo sapiens sont une longue durée de vie, grande taille du corps, une maturité sexuelle tardive, et une faible fécondité avec un fort investissement parental [50]. Une conséquence de ces traits d'histoire de vie est le potentiel de signaux environnementaux qui influencent la recherche de ressources, tant in utero, que chez l’adulte. Etant donné que l'inflammation est une réponse globale à de mauvaises conditions, une inflammation persistante peut signaler la présence d'un environnement défavorable et devrait conduire à un organisme à favoriser le maintien somatique sur la croissance et la reproduction (Figure 3B) [51]. Ainsi, la présence d'une inflammation chronique devrait affecter les traits d'histoire de vie à interpréter comme des coûts directs versus vulnérabilité.
La colite chronique fournit un bon exemple des effets que l'inflammation chronique peut avoir sur la croissance et la maturation compte tenu de son apparition pendant la phase de maturation [21]. Il a été montré que la maladie de Crohn est associée à un retard de croissance [52,53] et une puberté tardive [54,55], indépendante des apports alimentaires. Ces retards de croissance et de maturité sexuelle sont corrélés à un niveau élevé de sérum IL-6, et on a suggéré qu’ils pouvaient être lié à une suppression de l’insuline like growth factor 1 (IGF1) médiée par de l'IL-6 [56]. De meilleures évaluations des effets de l’élévation chronique des cytokines ont été réalisées chez des souris transgénique surexprimant IL-6. Ces souris étaient sensiblement plus petites que les souris normales [57] et les femelles étaient, soient infertiles, soit avec de très petites portées [58], confirmant que les cytokines inflammatoires peuvent modifier les traits d'histoire de vie. En outre, l'inflammation peut affecter la reproduction au cours même de la grossesse, comme contributeur majeur à la prématurité, aux retards de croissance intra-utérins, à la pré-éclampsie et aux avortements spontanés [59,60]. Enfin, chez les humains, la production de cytokines pro-inflammatoire en réponse à l'infection a été corrélée à une meilleure résistance aux infections tôt dans la vie, mais inversement corrélée avec la capacité globale de reproduction [61].
Pris ensemble, ces résultats suggèrent que les médiateurs de l’inflammation peuvent signaler certains aspects défavorables de l’environnement (comme l'abondance de pathogènes) et impacter directement la taille corporelle, les traits d'histoire de vie, l'âge de la reproduction, et la capacité globale de reproduction. Une prédiction de ce modèle est que l'inflammation peut favoriser le maintien somatique. On doit cependant noter, que, bien que tous les types de stress environnemental puissent promouvoir des programmes de maintenance somatiques au détriment de la croissance et de reproduction, différentes contraintes induisent différents types de programmes de maintenance. En outre, les différents programmes de maintenance peuvent être soit compatibles soit antagonistes l’un l'autre (figure 3C). Par exemple, si deux programmes de maintien reposent sur les mêmes ressources limitées, ils seront corrélés négativement ou reliés par un compromis. Par conséquent, l'inflammation, comme signal de stress environnemental, supprime non seulement la croissance et la reproduction pendant les périodes sensibles, mais elle peut aussi inhiber des programmes de maintenance antagonistes, tels que ceux qui contrôlent l'économie d'énergie. Les exemples simples de ce concept sont les troubles du comportement induits par l’infection, tels que l'anorexie, la fatigue chronique et les troubles du sommeil [21]. Si les troubles comportementaux peuvent favoriser les programmes de survie après l'infection [62], ils compromettent aussi les défenses contre la prédation, comme les réponses de lutte ou de fuite.
Perspectives: adaptations évolutives, l'inflammation et les maladies
Le point de vue évolutionniste discuté ci-dessus suggère que plusieurs facteurs peuvent contribuer à l'augmentation d'incidence des maladies inflammatoires.
Premièrement, le compromis coût-bénéfice des défenses inflammatoires a été optimisé pour un environnement qui n’existe plus pour la plupart des populations humaines, en particulier dans les pays industrialisés. L’adaptation à l'environnement opère par plusieurs mécanismes, dont l'adaptation génétique par sélection naturelle ; les réponses d’adaptation physiologiques et la plasticité phénotypique [63]. De plus, certains animaux peuvent modifier l'environnement à des degrés divers pour répondre à leurs besoins, processus connu sous le nom de ‘construction de niche’ [64]. Des exemples de construction de niche humaine comprennent l'agriculture, l'alimentation de type occidentale, l'habitat urbain, l'utilisation de vêtements, les produits d'hygiène, les antibiotiques et d'autres médicaments. Les êtres humains comptent sur la construction de niche plus que tout autre organisme, et cela est responsable des changements rapides et sans précédent de notre environnement moderne [65,66]. Cette rapide modification de multiples facteurs environnementaux, par la construction de niche, modifie à son tour le rapport coût-bénéfice optimal de nombreux traits, y compris la réponse inflammatoire (Figure 1). Par conséquent, les processus inflammatoires peuvent être mal engagés, ou dérégulés, provoquant une perturbation de l'homéostasie et favorisant certaines maladies, comme l’atopie, les maladies cardiovasculaires, l'obésité et le diabète de type 2.
Deuxièmement, l'inflammation est un trait à coût élevé et à haut bénéfice, ce qui la rend très vulnérables aux changements environnementaux qui peuvent inverser le ratio coûts-bénéfices (Figure 2). Compte tenu du nature coûteuse de la réponse inflammatoire, un équilibre coût-bénéfices sous optimal a un fort risque de diminuer la valeur sélective et d’augmenter le risque de maladie. En outre, les avantages de la réponse inflammatoire ont tendance à se manifester tôt dans la vie, quand ils ont le plus d'impact sur la reproduction, tandis que le «paiement» des coûts est souvent retardé jusqu'à le moment où l'organisme va, soit mourir de toutes façons, soit lorsque la maladie aura peu de conséquence sur la capacité de reproduction. Ce phénomène de pléiotropie antagonistes [67,68] est particulièrement pertinent pour des maladies inflammatoires chroniques qui se manifestent souvent à un âge avancé. L'augmentation de durée de la vie humaine dans les pays industrialisés, par réduction de la mortalité extrinsèque et amélioration des soins, entraîne, à son tour, une augmentation de la fraction de la population qui expérimente l'impact négatif de cette pléiotropie antagoniste.
Troisièmement, les coûts directs et les vulnérabilités de la plupart des caractères anatomiques et physiologiques humains peuvent affecter négativement l’homéostasie, en particulier lorsque ces traits ne sont pas optimisés pour un environnement changeant ou sont exposés à la pléiotropie antagoniste. Dans ce dernier cas, la valeur des coûts directs et la fréquence
des vulnérabilités augmente avec l'âge. Comme ces coûts augmentent, la perturbation de l'homéostasie augmente jusqu’à l’impossibilité d’être contrôlé par les mécanismes usuels, ce qui conduit à l'activation de ces mécanismes sur le mode inflammatoire. Conduisant ainsi à une inflammation chronique, puis modifiant les fonctions tissulaires et créant un cercle vicieux jusqu’aux pathologies inflammatoires communes : cardiovasculaires, métaboliques et neurodégénératives. Les maladies inflammatoires courantes sont des maladies de perturbation de l’homéostasie. Ainsi, les traits anatomiques et physiologiques de l'homme ne concernent pas seulement sur sa valeur sélective, mais agissent aussi sur les voies inflammatoires, créant des états inflammatoires chroniques qui déclencher, perpétuent ou aggravent les maladies de l'homéostasie.
Quatrièmement, l'inflammation peut favoriser les maladies à travers son effet sur les traits d'histoire de vie. L'inflammation est induite par des stimuli environnementaux nuisibles et peut donc signaler un environnement défavorable, une situation connue pour promouvoir les programmes de maintenance au détriment de la croissance et de la reproduction. Ainsi, l'inflammation peut entraîner certaines pathologies (retards de croissance, avortements spontanés) en raison des compromis fondamentaux intrinsèques aux traits d'histoire de vie. En outre, l'inflammation peut favoriser d’autres classes de maladies telles que les maladies métaboliques et autres maladies de l'homéostasie, en raison des interférences et incompatibilités entre programmes de maintenance et programmes inflammatoires.
En conclusion, une perspective évolutionniste peut fournir plusieurs indications sur la sensibilité humaine aux maladies inflammatoires, ainsi que sur l’augmentation de leur incidence dans les populations humaines modernes. Ironiquement, les progrès médicaux et la réduction de la mortalité extrinsèque par infections, famine ou prédation ont créé un nouvel ensemble de vulnérabilités aux maladies de l'abondance et de la vieillesse.
Cela est peut-être le plus grand de tous les compromis.
Traduction : Luc Perino)
Bibliographie
Okin D, Medzhitov R.
Evolution of Inflammatory Diseases Review.
Current Biology 22, R733–R740, September 11, 2012.
DOI: 10.1016/j.cub.2012.07.029
Références numérotées du texte
1. Hotamisligil, G.S., and Erbay, E. (2008). Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 8, 923–934.
2. Libby, P. (2006). Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nut. 83, 456S–460S.
3. Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35, 419–432.
4. Trinchieri, G. (2012). Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 30, 677–706.
5. Cecil, R.L., Goldman, L., and Ausiello, D.A. (2008). Medicine, 23rd edition (Philadelphia: Saunders Elsevier).
6. Nathan, C. (2002). Points of control in inflammation. Nature 420, 846–852.
7. Nathan, C., and Ding, A. (2010). Nonresolving inflammation. Cell 140, 871–882.
8. Murphy, K., Travers, P., Walport, M., and Janeway, C. (2008). Immunobiology, 7th edition (New York: Garland Science).
9. Medzhitov, R. (2010). Inflammation 2010: new adventures of an old flame. Cell 140, 771–776.
10. Kawai, T., and Akira, S. (2006). TLR signaling. Cell Death Diff. 13, 816–825.
11. Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832.
12. Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435.
13. Rocha e Silva, M. (1978). A brief survey of the history of inflammation. Agents Actions 8, 45–49.
14. Hotamisligil, G.S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867.
15. Zhang, K., Shen, X., Wu, J., Sakaki, K., Saunders, T., Rutkowski, D.T., Back, S.H., and Kaufman, R.J. (2006). Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599.
16. Zhang, K., and Kaufman, R.J. (2008). From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462.
17. Majno, G. (1975). The Healing Hand: Man and Wound in the Ancient World (Cambridge, Mass.: Harvard University Press).
18. Wynn, T.A. (2003). IL-13 effector functions. Annu. Rev. Immunol. 21, 425–456.
19. Gabay, C., and Kushner, I. (1999). Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454.
20. Rubanyi, G.M. (1993). The role of endothelium in cardiovascular homeostasis and diseases. J. Cardiovasc. Pharmacol. 22 (Suppl 4 ), S1–14.
21. Kumar, V., Abbas, A.K., Fausto, N., Robbins, S.L., and Cotran, R.S. (2005). Pathologic Basis of Disease, 7th edition (Philadelphia: Elsevier Saunders).
22. Blyth, D.I., Pedrick, M.S., Savage, T.J., Hessel, E.M., and Fattah, D. (1996). Lung inflammation and epithelial changes in a murine model of atopic asthma. Am. J. Resp. Cell Mol. Biol. 14, 425–438.
23. Lebreton, J.P., Joisel, F., Raoult, J.P., Lannuzel, B., Rogez, J.P., and Humbert, G. (1979). Serum concentration of human alpha 2 HS glycoprotein
during the inflammatory process: evidence that alpha 2 HS glycoprotein is a negative acute-phase reactant. J. Clin. Invest. 64, 1118–1129.
24. Gordon, A.H., and Koj, A. (1985). The Acute-Phase Response to Injury and Infection: The Roles of Interleukin I and Other Mediators (New York: Elsevier).
25. Kushner, I. (1993). Regulation of the acute phase response by cytokines. Perspect. Biol. Med. 36, 611–622.
26. Cannon, W.B. (1929). Organization for physiological homeostasis. Physiol. Rev. 9, 399–431.
27. Cannon, W.B. (1939). The Wisdom of the Body (New York: W.W. Norton & Company).
28. Pick, J. (1954). The evolution of homeostasis: The phylogenetic development of the regulation of bodily and mental activities by the autonomic nervous
system. Proc. Am. Philos. Soc. 98, 298–303.
29. Lin, H.V., and Accili, D. (2011). Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 14, 9–19.
30. Chrousos, G.P. (1995). The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332, 1351–1362.
31. Primrose, W.B. (1921). The evolution of the vertebrate endoskeleton: An essay on the significance and meaning of segmentation in coelomate animals. J. Anat. 55, 119–137.
32. Losos, J.B., Mason, K.A., Singer, S.R., Raven, P.H., and Johnson, G.B. (2008). Biology, 8th Edition (Boston: McGraw-Hill Higher Education).
33. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. (2000). Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum. 43, 1905–1915.
34. Falkowski, P.G., Katz, M.E., Milligan, A.J., Fennel, K., Cramer, B.S., Aubry, M.P., Berner, R.A., Novacek, M.J., and Zapol, W.M. (2005). The rise of oxygen over the past 205 million years and the evolution of large placental mammals. Science 309, 2202–2204.
35. Falkowski, P.G. (2006). Tracing oxygen’s imprint on earth’s metabolic evolution. Science 311, 1724–1725.
36. Hedges, S.B., Blair, J.E., Venturi, M.L., and Shoe, J.L. (2004). A molecular timescale of eukaryote evolution and the rise of complex multicellular life.
BMC Evol. Biol. 4, 2.
37. Becker, B.F. (1993). Towards the physiological-function of uric-acid. Free Radical Biol. Med. 14, 615–631.
38. Sedlak, T.W., Saleh, M., Higginson, D.S., Paul, B.D., Juluri, K.R., and Snyder, S.H. (2009). Bilirubin and glutathione have complementary antioxidant and
cytoprotective roles. Proc. Natl. Acad. Sci USA 106, 5171–5176.
39. Loeb, J.N. (1972). The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 15, 189–192.
40. Proctor, P. (1970). Similar functions of uric acid and ascorbate in man? Nature 228, 868.
41. Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Goutassociated uric acid crystals activate the NALP3 inflammasome. Nature 440,
237–241.
42. Conger, J.D. (1990). Acute uric acid nephropathy. Med. Clin. North Am. 74, 859–871.
43. Nathan, C. (2006). Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182.
44. Werner, S., and Grose, R. (2003). Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83, 835–870.
45. Parrillo, J.E. (1993). Pathogenetic mechanisms of septic shock. N. Engl. J. Med. 328, 1471–1477.
46. Dormandy, J.A. (1970). Clinical significance of blood viscosity. Ann. R. Coll. Surg. Eng. 47, 211–228.
47. Dormandy, J., and Hoare, E. (1972). Significance of raised blood viscosity in circulatory disease. Surg. Forum 23, 251–253.
48. Kamath, S., and Lip, G.Y. (2003). Fibrinogen: biochemistry, epidemiology and determinants. QJM-Mon. J. Assoc. Phys. 96, 711–729.
49. Stearns, S.C. (1992). The Evolution of Life Histories (Oxford, New York: Oxford University Press).
50. Hill, K., and Kaplan, H. (1999). Life history traits in humans: theory and empiricial studies. Annu. Rev. Anthropol. 28, 397–430.
51. Kopp, E.B., and Medzhitov, R. (2009). Infection and inflammation in somatic maintenance, growth and longevity. Evol. Appl. 2, 132–141.
52. Ballinger, A.B., Camacho-Hubner, C., and Croft, N.M. (2001). Growth failure and intestinal inflammation. QJM-Mon. J. Assoc. Phys. 94, 121–125.
53. Sawczenko, A., Azooz, O., Paraszczuk, J., Idestrom, M., Croft, N.M., Savage, M.O., Ballinger, A.B., and Sanderson, I.R. (2005). Intestinal inflammation induced growth retardation acts through IL-6 in rats and depends on the 174 IL-6 G/C polymorphism in children. Proc. Natl. Acad. Sci. USA 102, 13260–13265.
54. Azooz, O.G., Farthing, M.J.G., Savage, M.O., and Ballinger, A.B. (2001). Delayed puberty and response to testosterone in a rat model of colitis. Am. J. Physiol. Reg. I 281, R1483–R1491.
55. Ballinger, A.B., Savage, M.O., and Sanderson, I.R. (2003). Delayed puberty associated with inflammatory bowel disease. Pediatr. Res. 53, 205–210.
56. Ballinger, A.B., Azooz, O., El-Haj, T., Poole, S., and Farthing, M.J. (2000). Growth failure occurs through a decrease in insulin-like growth factor 1 which is independent of undernutrition in a rat model of colitis. Gut 46, 694–700.
57. De Benedetti, F., Alonzi, T., Moretta, A., Lazzaro, D., Costa, P., Poli, V., Martini, A., Ciliberto, G., and Fattori, E. (1997). Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. J. Clin. Invest. 99, 643–650.
58. De Benedetti, F., Pignatti, P., Vivarelli, M., Meazza, C., Ciliberto, G., Savino, R., and Martini, A. (2001). In vivo neutralization of human IL-6 (hIL-6) achieved by immunization of hIL-6-transgenic mice with a hIL-6 receptor antagonist. J. Immunol. 166, 4334–4340.
59. Jabbour, H.N., Sales, K.J., Catalano, R.D., and Norman, J.E. (2009). Inflammatory pathways in female reproductive health and disease. Reproduction
138, 903–919.
60. Challis, J.R., Lockwood, C.J., Myatt, L., Norman, J.E., Strauss, J.F., 3rd, and Petraglia, F. (2009). Inflammation and pregnancy. Reprod. Sci. 16, 206–215.
61. Van Den Biggelaar, A.H., De Craen, A.J., Gussekloo, J., Huizinga, T.W., Heijmans, B.T., Frolich, M., Kirkwood, T.B., and Westendorp, R.G. (2004). Inflammation underlying cardiovascular mortality is a late consequence of evolutionary programming. FASEB J. 18, 1022–1024.
62. Medzhitov, R., Schneider, D.S., and Soares, M.P. (2012). Disease tolerance as a defense strategy. Science 335, 936–941.
63. Stearns, S.C., and Hoekstra, R.F. (2005). Evolution: An Introduction, 2nd edition (Oxford, New York: Oxford University Press).
64. Laland, K.N., Odling-Smee, F.J., and Feldman, M.W. (1999). Evolutionary consequences of niche construction and their implications for ecology. Proc. Natl. Acad. Sci. USA 96, 10242–10247.
65. Laland, K.N., and Brown, G. (2006). Niche construction, human behavior, and the adaptive-lag hypothesis. Evol. Anthropol. 15, 95–104.
66. Rendell, L., Fogarty, L., and Laland, K.N. (2011). Runaway cultural niche construction. Phil. Trans. Roy. Soc. B Biol. Sci. 366, 823–835.
67. Medawar, P.B. (1952). An Unsolved Problem of Biology (London: Published for the college by H. K. Lewis).
68. Williams, G.C. (1957). Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411.
69. Hanahan, D., and Weinberg, R.A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
70. Finkel, T., and Holbrook, N.J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247.
71. Yu, B.P. (1994). Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 74, 139–162.
72. Levinson, D.J., and Sorensen, L.B. (1980). Renal handling of uric acid in normal and gouty subject: evidence for a 4-component system. Ann. Rheum.
Dis. 39, 173–179.
73. Choy, E.H., and Panayi, G.S. (2001). Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 344, 907–916.
74. Baskurt, O.K., and Meiselman, H.J. (2003). Blood rheology and hemodynamics. Semin. Throm. Hem. 29, 435–450.
75. Hord, N.G. (2008). Eukaryotic-microbiota crosstalk: p otential mechanisms for health benefits of prebiotics and probiotics. Annu. Rev. Nutr. 28, 215–231.
Et pour aller plus loin
Médecine évolutionniste (ou darwinienne)
Depuis quelques années, le problème de l'antibiorésistance, les progrès de la génomique, la redécouverte du microbiote et la prise en charge de maladies au long cours, nécessitent l'introduction d'une pensée évolutionniste dans la réflexion clinique.
Le premier diplôme universitaire intitulé "Biologie de l'évolution et médecine" a été mis en place à la faculté de Lyon en 2016.
Vous aimerez aussi ces humeurs...
Toucher des écrouelles - De tous temps, les souverains ont appuyé leur pouvoir sur la monnaie, l’armée, la justice, la [...]
Maladies en augmentation - Lorsque les épidémiologistes constatent l’augmentation de la prévalence d’une maladie, [...]
La probité à l'épreuve des certificats - Dans une société de droit et de normes, plusieurs professionnels ont la prérogative de [...]
Sexisme pittoresque de la médecine - Les médecins de l’ancienne Egypte considéraient l’hystérie féminine et les sorcelleries [...]
Comité de la fertilité - Tous les présidents de la Vème république ont insisté sur l'impératif d'alléger [...]
La phrase biomédicale aléatoire
La seule chose qui nous permettrait de donner notre accord à une théorie erronée serait l'absence d'une théorie meilleure ; de même, une injustice n'est tolérable que si elle est nécessaire pour éviter une plus grande injustice. Etant les vertus premières du comportement humain la vérité et la justice ne souffrent aucun compromis.
― John Rawls