
Médecine hamiltonienne : grands principes
dernière mise à jour le 10/01/2025
ATTENTION : Article pour experts de médecine évolutionniste !
Abstract
Nous présentons le domaine de la médecine hamiltonienne, qui se concentre sur les rôles de la parenté génétique dans la santé et les maladies humaines. La médecine hamiltonienne représente l'application de la théorie fondamentale de l'évolution sociale, pour les interactions impliquant la parenté, à des problèmes fondamentaux de la médecine tels que les agents pathogènes, le cancer, la croissance optimale et la maladie mentale. Elle englobe trois domaines, qui impliquent le conflit et la coopération entre : (i) les microbes ou les cellules cancéreuses, chez l'homme, (ii) les gènes exprimés chez l'homme, (iii) les individus humains. Un ensemble de six principes fondamentaux, basés sur ces domaines et leurs interfaces, sert à organiser conceptuellement le domaine et à contextualiser des exemples illustratifs. L'utilité principale de la médecine hamiltonienne est que, comme la médecine darwinienne plus généralement, elle fournit de nouvelles perspectives sur les données qu'il sera productif de collecter pour résoudre d'importants problèmes cliniques et de santé publique. Notre synthèse de ce domaine naissant est principalement destinée aux biologistes évolutionnistes et comportementaux qui aspirent à répondre à des questions directement liées à la santé et à la maladie humaines.
- Introduction
Le terme de médecine darwinienne fait référence à l'application de concepts et d'outils évolutionnistes pour comprendre la santé et les causes et traitements des maladies [ 1 ]. Ce domaine s'appuie sur le rôle de la sélection naturelle et d'autres processus évolutionnistes dans la médiation des risques et des symptômes des maladies. Dans ce contexte, la médecine darwinienne se concentre principalement sur les interfaces de maximisation de la valeur sélective, par les humains et leurs parasites, avec les écarts par rapport à la santé. La santé est considérée ici, selon la définition de l'Organisation mondiale de la santé, comme « un état de complet bien-être physique, mental et social et pas seulement la simple absence de maladie ou d'infirmité » [ 2 ]. Comme décrit ci-dessous, la correspondance de cet état avec les concepts évolutionnistes de fitness reste un sujet de recherche.
Les humains sont, bien sûr, censés maximiser non seulement leur fitness, mais aussi la fitness inclusive de Hamilton, qui intègre à la reproduction individuelle, celle d'autres individus qui peuvent porter les mêmes gènes par descendance [ 3 , 4 ]. Un phénotype est ainsi favorisé si les bénéfices ( b ) multipliés par la parenté génétique ( r ) avec un autre individu, moins les coûts ( c ) pour soi-même, sont supérieurs à zéro ( rb − c > 0), ce qui représente la célèbre règle de Hamilton. La fitness inclusive a été reconnue par les théoriciens et les praticiens de la médecine darwinienne comme une mesure fondamentale de la sélection naturelle [ 4 ]. Cependant, ses préceptes n'ont pas encore été systématiquement intégrés à la compréhension des bases évolutives de la santé et des maladies humaines.
Foster [ 5 ] a introduit le terme de médecine hamiltonienne à l'origine pour décrire les impacts sur les maladies infectieuses humaines de l'évolution induite par les interactions sociales entre les microbes. Dans cette revue, nous étendons et généralisons le concept de médecine hamiltonienne de Foster pour englober les rôles de la parenté génétique dans tous les aspects de la santé et des maladies humaines, infectieuses et non infectieuses. Ce faisant, nous décrivons un ensemble de principes centraux pour la médecine hamiltonienne, qui sont destinés à guider et à structurer son développement ultérieur. Nous soulignons que cette perspective sur la santé et la maladie se concentre sur des outils évolutionnaires théoriques et analytiques empiriquement bien fondés destinés à être appliqués à la médecine conventionnelle et traditionnelle, plutôt que de représenter une quelconque sorte d'approche alternative ou complémentaire.
Cette revue s'adresse principalement aux chercheurs en biologie évolutive et comportementale, qui pourraient être intéressés par des moyens d'étendre leurs intérêts et leurs études aux domaines de la médecine et de la santé. La définition et l'expansion de la médecine hamiltonienne en tant que domaine devraient cependant également fournir de nouvelles perspectives sur les processus fondamentaux de l'évolution, étant donné l'importance des humains et des microbes en tant que systèmes bien compris pour la recherche fondamentale.
- Domaines de la médecine hamiltonienne
La conception de Hamilton de la fitness inclusive affirme que les individus se comportent comme s'ils la maximisaient leur succès reproductif en y additionnant ceux de leur parentèle [ 6 ]. La médecine hamiltonienne, comme la théorie de la fitness inclusive, implique généralement une vision centrée sur les gènes des effets phénotypiques et de l'évolution, en insistant sur la parenté génétique. Les allèles exercent ainsi des effets phénotypiques qui modifient leurs taux de transmission, par rapport aux allèles alternatifs au même locus, par le biais d'effets de leurs copies chez les descendants ou les collatéraux.
Notre premier domaine de la médecine hamiltonienne correspond aux interactions entre cellules. Dans ce contexte, les interactions sociales entre microbes modulent les phénotypes microbiens qui affectent la santé de leurs hôtes humains. Ce domaine est celui des maladies infectieuses, car les microbes peuvent être transmis horizontalement entre hôtes humains. Ce domaine s'applique cependant également aux interactions sociales entre cellules cancéreuses au cours de l'évolution somatique de la carcinogenèse.
Les analyses au niveau cellulaire examinent pourquoi et comment les cellules coopèrent et rivalisent en fonction de la parenté génétique, des coûts et des avantages. La coopération et la compétition affectent directement la transmission microbienne, la virulence et la résistance aux défenses de l'hôte. La transmission microbienne, la virulence et les adaptations liées à la maladie dépendent en outre de la structure de la population de leurs hôtes humains, car les réponses individuelles à l'infection influencent les risques de maladie chez les parents [ 1 ]. Parmi les cellules cancéreuses, la coopération et la compétition basées sur la parenté modulent également le comportement cellulaire et la résistance à la chimiothérapie.
Le deuxième domaine correspond aux interactions entre gènes. Dans ce contexte, les gènes d'un même individu peuvent entrer en compétition ou coopérer entre eux, en fonction de leur parenté génétique avec des partenaires sociaux. Les différences entre les gènes en termes de parenté avec les interlocuteurs peuvent être causées par une variation dans la transmission des gènes (en particulier, un gène transmis à un sexe spécifique ou exprimé lorsqu'il est hérité d'un sexe spécifique) ou dans la correspondance des gènes (par exemple, les barbes vertes [ 4 ]). Comme décrit plus en détail ci-dessous, le conflit intragénomique potentialise les risques de maladie et peut amplifier les impacts sur la santé de la dysrégulation physiologique.
Notre troisième domaine correspond aux interactions entre humains. Dans ce contexte, les interactions sociales interhumaines modulent la santé et la maladie humaines. Les analyses à ce niveau examinent pourquoi et comment les individus rivalisent et coopèrent en fonction de la parenté génétique avec leurs interlocuteurs sociaux. Un tel conflit intergénomique est attendu chaque fois que la parenté entre un acteur et un destinataire est inférieure à un, ce qui est presque toujours le cas. En raison de ces différences de parenté, les phénotypes optimaux pour une paire d'individus en interaction divergent, ce qui peut entraîner un risque accru de maladie pour l'un d'eux ou pour les deux.
Ces trois domaines délimitent le champ d'application global de la médecine hamiltonienne. Nous décrivons ensuite une série de principes fondamentaux pour ce domaine, en faisant référence à des concepts et à une théorie pertinents, à des exemples illustratifs et à une réflexion sur la manière dont le domaine peut être encore amélioré.
- Six principes de la médecine hamiltonienne
(a) La parenté entre les microbes influence leur virulence, leur dynamique de transmission et d'autres effets sur les hôtes
Les microbes pathogènes présentent un large éventail d’interactions sociales coopératives qui influencent leurs moyens et leurs capacités à exploiter les hôtes humains [ 7 – 9 ] ( tableau 1 ). Une telle coopération implique normalement une parenté génétique non nulle, selon laquelle les cellules interagissant de manière altruiste ou mutualiste ont tendance à partager un allèle associé à un certain comportement, généralement à la suite d’une descendance clonale. Par exemple, il existe de nombreux exemples chez les microbes de cellules individuelles d’un clone qui se sacrifient pour améliorer la survie et la réplication des clones, ce qui augmente vraisemblablement la fréquence relative de l’allèle altruiste et du clone dans les générations futures [ 7 , 8 ].
Tableau 1.
|
Le comportement social microbien et sa pertinence pour la santé humaine et les maladies infectieuses. |
||||||
|
|
||||||
|
Comportement social microbien |
|
Pertinence pour la santé |
|
Pertinence par rapport au traitement |
|
réf |
|
Sécrétion altruiste ou malveillante de bactériocines pour tuer les souches concurrentes |
|
Les sécrétions interviennent directement dans la dynamique des populations microbiennes |
|
Développer des bactériocines en tant qu’antibiotiques à spectre étroit ou large |
|
[10,11] |
|
Dormance altruiste, qui épargne les ressources pour les parents |
|
La dormance peut compliquer l’élimination des infections par les antibiotiques |
|
Manipuler les systèmes de dormance |
|
[8,12] |
|
Discrimination par la parenté |
|
La discrimination permet l’ajustement conditionnel du comportement en ce qui concerne la parenté |
|
Manipuler les systèmes de discrimination pour réduire la virulence |
|
|
|
Une croissance plus efficace et plus rapide avec une plus grande parenté |
|
La virulence varie positivement avec la parenté microbienne dans une infection |
|
Augmenter la diversité génotypique d’une infection ; manipuler les indices de parenté |
|
|
|
Utilisation prudente des ressources dans le cadre d’un compromis taux-efficacité et d’une forte corrélation |
|
La prudence peut augmenter ou diminuer la virulence ou la persistance chronique |
|
Manipuler les mécanismes de compromis |
|
|
|
Production de biofilms |
|
Les biofilms protègent les microbes des environnements difficiles, améliorent la croissance et la persistance |
|
Développer des moyens de perturber la production et la maintenance des biofilms |
|
[14,15] |
|
Transfert horizontal forcé de gènes du facteur de virulence |
|
Les transferts augmentent la virulence |
|
Manipuler les mécanismes de transfert |
|
[16 à 18] |
|
La communication conjointe de biens publics ; Exploitation des tricheurs |
|
Les biens publics favorisent la virulence |
|
Inoculer des souches de tricheurs ou des facteurs qui favorisent les tricheurs / Cibler les biens publics plutôt que les microbes |
|
[19 à 22] |
|
Suicide altruiste de microbes infectés par des phages |
|
Le suicide favorise la survie des clones |
|
Manipuler les indices de suicide |
|
[23 à 25] |
|
Détection du quorum via des molécules de signalisation diffusibles |
|
La communication intercellulaire améliore la régulation de la croissance et de la virulence |
|
Manipuler ('quench') des systèmes de détection de quorum |
|
[26 à 29] |
Comme chez les humains, la coopération entre microbes implique généralement une ressource générée socialement, comme des produits génétiques partagés et sécrétés, qui peuvent être exploités par des génotypes « tricheurs » qui récoltent les bénéfices de la coopération mais évitent ses coûts. Cette tricherie, à son tour, sélectionne des mécanismes qui limitent les pertes liées à l'exploitation, comme la répression des tricheurs, la pléiotropie entre les traits coopératifs et fonctionnellement essentiels, et la régulation hautement conditionnelle des phénotypes coopératifs [ 9 , 30 , 31 ]. Ces comportements microbiens modulent les maladies infectieuses humaines, car ces traits sociaux et antisociaux affectent directement la dynamique des populations microbiennes, la virulence, la transmission et l'efficacité des stratégies humaines visant à réduire les impacts des maladies infectieuses sur la santé [ 7 , 8 ].
La virulence des maladies infectieuses, un déterminant majeur des effets sur la santé, peut être associée à la parenté génétique intra-hôte microbienne d'au moins trois façons différentes, selon la théorie actuelle. Premièrement, la virulence peut être inversement associée à la parenté génétique, si les génotypes microbiens rivalisent plus fortement pour exploiter les ressources de l'hôte en raison des interactions intra-hôte avec des non-parents [ 32 – 34 ]. Alternativement, la virulence peut être plus élevée avec une parenté plus étroite, lorsque la coopération entre microbes apparentés augmente leur capacité à se reproduire rapidement et à mieux exploiter les hôtes humains [ 35 ]. Troisièmement, la relation entre virulence et parenté peut dans certaines circonstances être en forme de U, avec une virulence plus faible sous des niveaux intermédiaires de parenté [ 10 ]. La réalisation de ces attentes alternatives dans les populations naturelles devrait dépendre, en partie, des mécanismes de coopération et de compétition, ou de tricherie, pour toute interaction donnée microbe-hôte, qui déterminent comment les comportements microbiens affectent leur dynamique de population [ 34 ]. Par exemple, une parenté plus élevée peut favoriser la virulence lorsque la réplication microbienne au sein d'un hôte dépend plus fortement de la coopération, mais une parenté plus faible peut favoriser la virulence sous sélection pour une utilisation plus rapide, bien que moins efficace, des ressources de l'hôte. De plus, lorsqu'une parenté plus faible est liée à une fréquence plus élevée de tricherie, la virulence peut être une fonction dépendante de la fréquence des coopérateurs et des tricheurs [ 36 ]. Comme prévu, compte tenu de ces considérations, certains microbes présentent des adaptations interprétables comme la reconnaissance de clones [ 9 , 18 , 37 ] ; chez d'autres espèces, le comportement adaptatif lié à la virulence peut être associé à des niveaux de parenté à long terme typiques de la population au sein des hôtes, sans reconnaissance de parenté en soi .
L'extension et la vérification de la théorie de la fitness inclusive pour l'évolution de la virulence, ainsi que l'élucidation des différentes conditions comportementales, écologiques et évolutives qui produisent différentes relations entre la virulence et la parenté génétique, restent des défis majeurs pour les étudiants de la socialité microbienne [ 35 , 38 , 39 ]. Comme pour les créatures sociales macroscopiques, le contrôle des phénotypes sociaux et la spécification de la parenté, essentiels à l'application de la règle de Hamilton, revêtent une importance capitale. Par exemple, les cellules microbiennes s'engagent couramment dans un transfert horizontal de gènes de plasmides ou d'autres éléments qui codent des facteurs de virulence [ 18 ] ; de tels transferts peuvent être sous le contrôle de gènes dans l'ADN transmis et peuvent contraindre les cellules réceptrices à la coopération sociale [ 16 , 17 ]. De tels phénomènes représentent également la transmission sociale de la parenté elle-même, car les microbes deviennent génétiquement liés via un transfert horizontal, pour le locus considéré [ 17 , 40 ]. La règle de Hamilton peut également être plus compliquée chez les microbes en raison d'une incidence élevée d'effets de sélection forts et non additifs, ce qui nécessite des ajustements à rb − c > 0 pour la prédiction des phénotypes comportementaux [ 41 ]. Enfin, le comportement social et la virulence microbienne dépendent de manière cruciale des échelles spatiales des interactions compétitives et coopératives entre parents, étant donné que les microbes vivent dans des groupes denses et écologiquement interactifs [ 42 ] ; la compétition entre parents peut ainsi supprimer l'évolution de la coopération dans certaines conditions ou sélectionner des traits microbiens, tels que les cellules « résistantes » quiescentes ou la mort cellulaire programmée, qui réduisent ses effets [ 8 , 9 ].
Jusqu'à présent, les études médicales hamiltoniennes sur les microbes se sont concentrées sur les bactéries pathogènes et sur des considérations issues de la théorie de l'évolution plutôt que sur l'écologie. Cependant, les microbes pathogènes sont généralement des parents phylogénétiques proches des microbes commensaux ou mutualistes humains [ 43 ], les microbes pathogènes et mutualistes existent fréquemment le long de continuums même au sein d'«espèces» microbiennes nominales [ 43 , 44 ], et les composantes mutualistes du microbiome humain semblent exercer des impacts sur la santé au moins aussi puissants que ceux des agents pathogènes [ 45 ]. Les considérations issues de la médecine hamiltonienne peuvent-elles aider à expliquer la coopération et la compétition chez les microbes humains mutualistes, et les transitions entre mutualisme et pathogénicité dans différents contextes écologiques et évolutifs [ 44 ] ? Comment les rétroactions écologiques-évolutives, qui devraient être particulièrement fortes chez les bactéries, médiatisent-elles les phénotypes bactériens sociaux [ 46 , 47 ] ? Dans quelle mesure et de quelle manière la compétition et la coopération entre les virus influencent-elles leur virulence et leur transmission [ 48 , 49 ] ? Le tableau 1 résume les comportements sociaux microbiens, leur pertinence pour la santé et leur utilité potentielle pour la prévention et le contrôle des maladies infectieuses, afin de motiver de nouveaux progrès dans ce domaine clé de la médecine hamiltonienne.
(b) La structure de parenté humaine sert de médiateur aux symptômes, à la virulence et à la transmission des agents pathogènes et des mécanismes de défense
L'idée clé de Hamilton pour comprendre l'évolution sociale est que la parenté génétique influence le comportement lié à la valeur sélective de manière mathématiquement prévisible. Pour analyser la santé d'un point de vue hamiltonien, il est essentiel de reconnaître que le « comportement » comprend tous les phénotypes liés à la maladie ayant des effets sur la parenté, qu'il s'agisse de défenses ou de réponses immunitaires, de symptômes de maladie, d'interactions sociales qui modulent la transmission ou de modèles différentiels d'investissement et de compromis concernant la croissance, l'entretien et la reproduction.
De même que la parenté microbienne affecte la santé humaine, la parenté humaine affecte également les risques, la transmission et les symptômes des maladies infectieuses. La parenté humaine influence la maladie car dans les groupes structurés par la parenté, les fonctions du système immunitaire, les phénotypes de maladies exprimés par l'hôte et les adaptations des hôtes pour éviter, résister ou combattre la maladie représentent des traits sociaux avec des effets importants attendus sur les parents [ 32 – 34 , 50 ]. Les adaptations de l'hôte pour réduire les effets délétères des maladies infectieuses comprennent la résistance (éviter l'infection ou investir dans des réponses immunitaires pour l'éliminer de l'organisme) et la tolérance (atténuer les effets néfastes de l'infection, sans élimination). Frank [ 32 ] a utilisé un modèle d'aptitude inclusive pour montrer qu'une parenté plus élevée sélectionne un investissement accru dans la résistance (éviter l'infection, dans ce cas, par l'induction de l'immunité). De telles stratégies de résistance peuvent inclure des réponses et des comportements altruistes liés à la maladie des hôtes humains, qui réduisent la transmission du pathogène aux parents [ 25 , 51 , 52 ]. Il est également prévu que la structure de parenté limite l'évolution de la tolérance [ 51 , 52 ], en partie parce que les individus tolérants restent contagieux. De tels effets peuvent être particulièrement importants en raison des compromis généralisés entre l'investissement dans les fonctions immunitaires et l'investissement dans la croissance et la reproduction, les deux contextes habituels pour envisager la maximisation de la fitness inclusive.
Best et al . [ 53 ] ont corroboré les résultats de Frank et ont également montré, à l'aide d'un modèle co-évolutionniste, que des interactions plus locales entre membres d'une même famille favorisent une transmissibilité et une virulence réduites des parasites. Ces résultats sont importants car leur modèle prédit qu'un mélange accru, tel qu'il est observé dans les populations actuelles, favoriserait une combinaison de virulence et de transmissibilité élevées des agents pathogènes avec une résistance réduite de l'hôte à l'infection, précisément les conditions qui favorisent les pandémies humaines avec des taux de mortalité élevés.
La dynamique de transmission des maladies et l'évolution des allèles qui influencent la résistance aux maladies sont également influencées par la parenté, car les allèles qui protègent un individu contre la maladie protégeront également les membres de sa famille qui partagent des copies de gènes . Schliekelman [ 54 ] a montré que la force de la sélection pour la résistance dépend de manière cruciale de la manière dont les allèles de résistance influencent les taux de transmission interfamiliale par rapport à la transmission intrafamiliale. Ces taux ont été mesurés, démontrant les effets importants de la structure de parenté ; par exemple, la transmission de Yersinia pestis (peste) entre ménages en Angleterre à la fin du XVIIe siècle était « généralement réalisée par des visites à des proches parents » [ 55 , p. 135].
Enfin, Williams et Nesse [ 1 ] ont souligné que les symptômes de maladies qui semblent représenter des manipulations des hôtes par des agents pathogènes, comme la toux et les éternuements, peuvent présenter de grands avantages pour les agents pathogènes mais de faibles coûts pour les hôtes, à moins que ces symptômes n'aient un impact différent sur les parents de l'hôte. Ces considérations indiquent que les effets d'adaptation inclusive peuvent influencer tous les phénotypes liés aux maladies humaines qui modifient les taux de transmission, car les humains vivent dans des groupes structurés par la parenté. En ce qui concerne les situations de conflit au sein des espèces, les résultats des conflits entre l'agent pathogène et l'hôte sur les symptômes de la maladie qui affectent la transmission dépendent des forces de sélection des deux parties et des stratégies phénotypiques dont elles disposent ; dans ce contexte, dans quelle mesure les symptômes de la maladie représentent-ils des effets de la sélection impliquant des parents ?
L'étude des effets de parenté chez les microbes pathogènes offre des opportunités exceptionnelles aux biologistes évolutionnistes, aux écologistes et aux biologistes comportementaux qui souhaitent se former en microbiologie, en biologie moléculaire ou en épidémiologie. Les recherches menées dans plusieurs groupes [ 8 , 9 ] constituent des exemples de réussite dans de telles études interdisciplinaires.
(c) Les effets de l'aptitude inclusive favorisent la croissance et la résistance au traitement des cellules cancéreuses
Les applications des perspectives socio-comportementales et de la médecine évolutionniste à l'étude de la carcinogenèse se sont jusqu'à présent concentrées principalement sur trois domaines : (i) la sélection naturelle entre des lignées de cellules cancéreuses génétiquement divergentes pour des phénotypes qui augmentent leur taux de réplication, (ii) la compétition des cellules cancéreuses avec les cellules normales et (iii) la coopération entre les cellules cancéreuses, qui, selon certaines hypothèses, génère des « sociétés de cellules tumorales » [ 56 – 60 ]. De telles études ont mis en évidence les causes et les effets de l'hétérogénéité génétique entre les cellules d'une tumeur, ainsi que les mécanismes moléculaires par lesquels les cellules interagissent les unes avec les autres de manière positive ou négative, mais les considérations explicites de la théorie de la fitness inclusive n'ont, étonnamment, pas encore été appliquées.
La parenté génétique entre les cellules cancéreuses est essentiellement la même que pour les groupes de microbes : une cellule qui abrite un allèle qui contrôle un phénotype est apparentée par 1 à d'autres cellules réceptrices ayant le même allèle (identique par descendance ou transfert horizontal [ 61 ]), et par 0 à toute cellule donnée ayant un allèle alternatif. Dans les groupes locaux de cellules à génotypes mixtes, la parenté est comprise entre 0 et 1, car les effets de l'allèle d'une cellule focale ont un impact sur les cellules qui le portent, et sur celles qui ne le portent pas.
Comment les cellules cancéreuses plus ou moins apparentées interagissent-elles, au sens comportemental ? Comme les microbes, elles expriment des molécules à la surface cellulaire et sécrètent des produits diffusibles (biens publics), qui modulent le comportement (modèles d'expression génétique et phénotypes interactifs) des cellules voisines, ainsi que leur environnement [ 62 – 65 ]. Les cellules cancéreuses forment également, comme les microbes, des structures tridimensionnelles hétérogènes, composées de mélanges de cellules qui diffèrent en génotype et en phénotype [ 56 ]. Ces cellules peuvent différer phénotypiquement les unes des autres de manière socialement pertinente : par exemple, les tumeurs restent très petites jusqu'à ce que certaines cellules subissent un « changement angiogénique » pour se spécialiser dans le développement de tissus qui servent de vaisseaux sanguins, fournissent des nutriments et éliminent les déchets [ 60 ]. Cette variation morphogénétique semble représenter une forme de division cellulaire du travail impliquant des compromis entre la prolifération et l'approvisionnement en nourriture, l'angiogenèse étant un phénotype altruiste qui devrait être sensible à l'invasion secondaire de clones « tricheurs » qui exploitent les ressources fournies [ 60 ]. En effet, « le potentiel angiogénique repose sur la production coopérative du signal angiogénique » [ 60 ], de la même manière que les produits partagés et sécrétés médiatisent de nombreuses interactions sociales entre microbes pathogènes.
Nagy [ 66 , 67 ] a utilisé des modèles mathématiques de compétition entre les types de cellules cancéreuses, en relation avec les degrés de vascularisation et la croissance tumorale globale, pour analyser la dynamique attendue de ces compromis. Ses modèles ont démontré que la compétition entre les lignées de cellules cancéreuses pouvait conduire au développement d' «hypertumeurs» hautement prolifératives, qui surpassent et détruisent les tissus cancéreux préexistants, puis s'autodétruisent en raison de leur manque de capacité d'angiogenèse, ou persistent avec d'autres tissus tumoraux à un équilibre dépendant de la fréquence. L'évolution somatique des hypertumeurs est cohérente avec les exemples de régression spontanée et de zones de nécrose dans le neuroblastome et d'autres cancers [ 66 ], mais elle reste à étudier en détail.
Les hypertumeurs représentent une trajectoire possible d'interactions au sein et entre les génotypes et les lignées de cellules cancéreuses ; vraisemblablement, d'autres phénotypes comportementaux cellulaires compétitifs et coopératifs moins spectaculaires modulent la carcinogenèse depuis son apparition jusqu'à sa régression ou sa malignité. Un autre exemple potentiel concerne les avantages procurés aux cellules souches cancéreuses par des cellules clonalement apparentées et partiellement différenciées (les « aides au nid » des cellules cancéreuses, en quelque sorte) [ 68 ] ; ce processus a été démontré dans des simulations [ 69 ] et peut être d'une importance considérable pour le comportement et l'évolution des cellules cancéreuses.
Prises ensemble, ces considérations suggèrent que l'évolution des cellules cancéreuses ressemble fondamentalement à celle des microbes ( tableau 2 ), malgré le fait que l'évolution des cellules cancéreuses se déroule de novo dans chaque cas de carcinogenèse et implique une cooptation somatique-évolutive de gènes et de voies qui ont évolué dans le contexte d'activités cellulaires normales. Les cancers transmissibles chez les chiens domestiques et les diables de Tasmanie [ 78 ] représentent en effet des ponts empiriques entre les maladies infectieuses et la carcinogenèse, tout comme l'application de méthodes expérimentales-évolutives [ 79 ] et le développement d'agents thérapeutiques relativement « à l'épreuve de l'évolution » qui retardent ou empêchent l'évolution de la résistance [ 80 ]. Les microbes eux-mêmes peuvent même être soumis à une prolifération cancéreuse par des « lignées rebelles » [ 81 ], dont le contrôle et la dynamique devraient fournir des informations sur la répression des cellules surproliférantes de manière beaucoup plus générale [ 81 , 82 ].
Tableau 2.
|
Parallèles entre les cellules cancéreuses et les cellules microbiennes, en ce qui concerne les comportements sociaux liés à la parenté qui ont un impact sur la santé humaine. |
||||
|
|
|
|
|
|
|
Comportement social des cellules cancéreuses |
|
Parallèle microbien |
|
Réf |
|
Sécrétion cellulaire de substances chimiques qui modifient la survie, la croissance et le comportement d'autres cellules (clones, autres cellules cancéreuses et cellules normales) |
|
Bactériocines, biens publics, signalisation chimique |
|
[ 59 , 64 , 65 , 70 ] |
|
Modulation par détection de quorum des modèles de croissance cellulaire, de quiescence, de dispersion et d'immortalisation des cellules souches |
|
Détection de quorum qui module la croissance, le comportement et la virulence |
|
[ 63 , 71 ] |
|
Différenciation de certaines cellules en cellules « auxiliaires » moins reproductrices (par exemple, le commutateur angiogénique) |
|
Cellules microbiennes différenciées et moins reproductrices |
|
|
|
Coopération intercellulaire mutualiste et altruiste, généralement au sein de clones |
|
Formes diverses |
|
[ 57 , 72 ] |
|
Interactions mutualistes et altruistes dans des groupes de cellules qui expriment des phénotypes au niveau du groupe ; les groupes peuvent comprendre des clones ou des mélanges clonaux |
|
Phénotypes multicellulaires chez de nombreuses bactéries |
|
[ 56 , 60 , 63 , 73 , |
|
Réponses coopératives au stress entre les cellules en interaction, favorisant l'évolution de la résistance aux médicaments |
|
Evolution sociale de la résistance aux antibiotiques |
|
|
|
Transfert horizontal de gènes, médiation de la survie et de la croissance cellulaire |
|
Transfert horizontal de plasmides et d'autres sources d'ADN |
|
[ 61 , 76 , 77 ] |
|
Dispersion altruiste, par laquelle la dispersion de certaines cellules libère des ressources pour les clones (?) |
|
Peut fonctionner dans le contexte des biofilms, d'autres groupements |
|
n / A |
|
Suicide altruiste, par lequel la mort cellulaire programmée libère des ressources pour les clones dans des conditions stressantes (?) |
|
Signalé chez certaines bactéries |
|
n / A |
La conceptualisation et la modélisation de la dynamique des populations de cellules cancéreuses en termes de théorie de la fitness inclusive et de comportement coopératif et compétitif basé sur la parenté devraient conduire à de nouvelles perspectives ayant des implications directes pour la thérapie. Les chercheurs à l'échelle de l'organisme en ce domaine devront investir massivement dans l'apprentissage de la biologie du cancer, et les recherches ayant les plus forts impacts impliqueront probablement des travaux empiriques menés par des équipes intégratives.
(d) Les conflits intragénomiques influencent les phénotypes et les risques de maladie
Le conflit intragénomique découle directement de la vision hamiltonienne de l'évolution [ 83 ]. Par ce processus, la divergence des intérêts génétiques est causée par des différences entre des ensembles de gènes dans le modèle de transmission ou dans la correspondance des gènes (via les gènes à barbe verte), ce qui entraîne une parenté différentielle des gènes focaux avec les interlocuteurs sociaux de leurs porteurs [ 4 ]. Par exemple, les femelles de la même fratrie chez les mammifères euthériens sont apparentées à 3/4 pour les gènes liés au chromosome X, mais à 1/2 pour les gènes autosomiques ; de telles factions génomiques [ 84 ] peuvent ainsi être en conflit sur un phénotype si rb − c > 0 pour une partie mais pas pour l'autre. Plus généralement, les gènes codant pour un phénotype particulier sont en conflit lorsque la valeur phénotypique qui maximise la réplication d'un gène est différente du phénotype qui maximise la réplication de l'autre gène (différemment hérité ou apparenté). Par ce processus, les allèles qui font progresser leur propre représentation dans les générations futures peuvent, dans certaines circonstances, augmenter leur prévalence même s’ils augmentent les risques de maladie.
Les différences de parenté peuvent conduire les génomes individuels à montrer des signes de conflit entre les gènes autosomiques, chromosomiques sexuels et mitochondriaux ( figure 1 ). Prenons par exemple l'altruisme envers les sœurs. Les gènes mitochondriaux favorisent le plus haut niveau d'altruisme, tandis que les gènes du chromosome Y favorisent le plus bas. Un conflit peut également exister entre les gènes hérités de la mère, les gènes hérités du père et les gènes qui ne portent pas d'informations sur leur origine parentale. Prenons par exemple l'altruisme envers les frères et sœurs matrilinéaires. Les gènes hérités de la mère favorisent le plus haut niveau d'altruisme, tandis que les gènes hérités du père favorisent le plus bas. Enfin, les génomes peuvent montrer des signes de conflit entre un allèle spécifique qui se reconnaît dans un partenaire social (un « gène de la barbe verte ») et un allèle qui ne se reconnait pas [ 4 , 85 , 86 ].
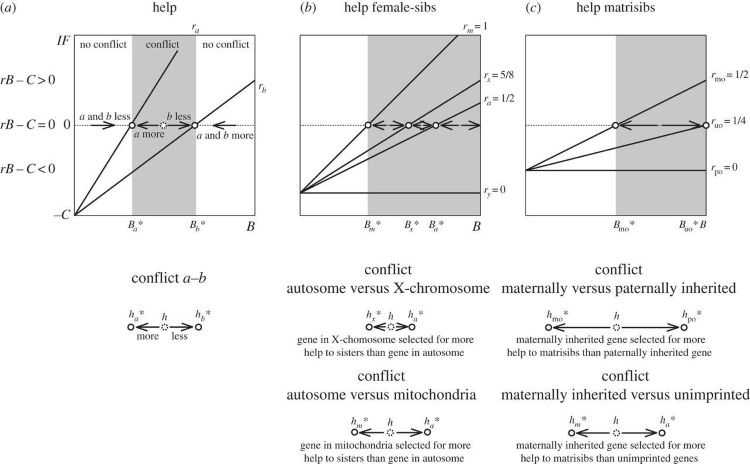
Figure 1 : Conflit entre les gènes sous-jacents au comportement d'aide.
L' axe des x représente le bénéfice ressenti par le bénéficiaire de l'aide ( B ), tandis que l' axe des y représente la fitness inclusive de l'acteur ( IF ). La stratégie évolutivement stable correspond au cas IF = rB – C = 0, où C est le coût subi par l'acteur et r est le coefficient de parenté. Cela correspond graphiquement au point où la ligne IF coupe la ligne 0. Lorsque IF < 0, un gène est sélectionné pour moins d'aide, lorsque IF > 0, un gène est sélectionné pour plus d'aide. ( a ) Cette figure correspond à deux gènes génériques, a et b , codant pour l'aide. Les gènes sont soumis à une pression sélective différente lorsque leur parenté avec les bénéficiaires diffère, c'est-à-dire r a ≠ r b . Lorsque la parenté du gène a est supérieure à celle du gène b , c'est-à-dire r a > r b , le bénéfice optimal pour le gène a est inférieur à celui du gène b , c'est-à-direet le gène a est sélectionné pour plus d'aide que le gène b . La région où les gènes a et b sont sélectionnés pour une aide différente est une région de conflit (en gris sur les figures). ( b ) Cette figure correspond aux gènes codant pour une aide dirigée vers les sœurs de sexe féminin. Les gènes mitochondriaux ( m ) sont sélectionnés pour plus d'aide que les gènes liés à l'X ( x ). Les gènes liés à l'X sont sélectionnés pour plus d'aide que les gènes autosomiques ( a ). Les gènes autosomiques sont sélectionnés pour plus d'aide que les gènes liés à l'Y ( y ). Par conséquent, en fonction du niveau d'aide actuel, il peut y avoir un conflit entre n'importe laquelle de ces paires de gènes, à savoir : m versus x , x versus a , a versus y , m versus a , m versus y , x versus y . ( c ) Cette figure correspond aux gènes codant pour une aide dirigée vers les sœurs maternelles. Les gènes d'origine maternelle (mo) sont sélectionnés pour plus d'aide que ceux d'origine inconnue (uo). Les gènes d'origine inconnue sont sélectionnés pour une aide plus importante que ceux d'origine paternelle (po). Par conséquent, en fonction du niveau d'aide actuel, il peut y avoir un conflit entre l'une de ces paires de gènes, à savoir : mo versus uo, uo versus po, mo versus po.
Dans le contexte des conflits intragénomiques, le génome peut être considéré comme un système social de gènes et de voies coopérants et concurrents, mais mutuellement dépendants, comprenant différentes factions selon leur localisation autosomique, chromosomique sexuelle ou cytoplasmique et leur expression paternelle ou maternelle [ 84 , 87 ]. Les impacts sur la santé et la maladie découlent directement des effets du comportement génique résultant sur la forme physique relative.
L'issue ou la résolution d'un conflit intragénomique devrait dépendre de l'ensemble de gènes qui contrôle le phénotype résultant. Par exemple, le contrôle des valeurs phénotypiques devrait être influencé par le nombre de gènes inclus dans chaque ensemble [ 4 ]. Pour cette raison, de nombreux cas de conflit entre gènes autosomiques et gènes chromosomiques sexuels ou mitochondriaux devraient, en théorie, être résolus à l'avantage des gènes autosomiques [ 87 , 88 ]. En revanche, le nombre de gènes hérités de la mère et du père sera presque également réparti (également chez les femmes, mais inégalement chez les hommes qui ont un nombre légèrement plus élevé de gènes hérités de la mère) et donc la résolution de tels conflits dépendra du phénotype spécifique codé et des détails des interactions moléculaires.
Les conflits intragénomiques peuvent accroître les risques de maladie de plusieurs façons. Tout d’abord, on s’attend à ce que ces conflits entraînent l’évolution de nouveaux mécanismes de régulation génétique et épigénétique liés aux conflits. La complexité accrue de ces mécanismes de régulation peut augmenter la probabilité de dysrégulation, par rapport aux situations non conflictuelles [ 89 – 91 ].
Deuxièmement, le conflit intragénomique peut provoquer une décanalisation des trajectoires et des fonctions développementales [ 92 ]. Par ce mécanisme, tout degré de conflit entraîne une expression accrue des gènes antagonistes ou une sélection en faveur d'autres moyens de « gagner ». La mesure dans laquelle cette escalade se poursuit n'est pas déterminée par l'ampleur du conflit, mais par les coûts de l'expression accrue de chaque gène antagoniste. En conséquence, une perturbation des équilibres dynamiques établis par les gènes en conflit intragénomique tend à entraîner des pathologies plus souvent et plus extrêmes que les perturbations des gènes non en conflit [ 88 , 93 ]. Le syndrome de sous-croissance de Silver-Russell et le syndrome de surcroissance de Beckwith-Wiedemann représentent des exemples paradigmatiques de pathologies causées par la dysrégulation des systèmes de conflit de gènes imprimés, ici médiée principalement par des altérations des domaines imprimés IGF2/H19 et CDKN1C/KCNQ1OT1 [ 94 , 95 ].
Troisièmement, les conflits intragénomiques devraient impliquer une sélection antagoniste pour des effets aussi tôt que possible dans le développement, et une sélection pour le contrôle de la dynamique des cellules souches. De tels effets sont attendus car au cours du développement, le pouvoir sur les phénotypes est généralement (i) une fonction du temps, car les événements plus tôt dans l’ontogenèse ont des effets plus importants et plus en cascade, et (ii) une fonction de contrôle sur les taux et les modèles de prolifération cellulaire et tissulaire, car c’est ainsi que les organismes grandissent et se différencient. Cette prédiction est bien étayée par des études récentes montrant que dans une large gamme de tissus humains, les gènes imprimés orchestrent la réplication des cellules souches plutôt que leur inhibition [ 96 – 98 ], et par l’observation que les gènes imprimés exercent des effets particulièrement répandus sur le développement placentaire, au début de la croissance et du développement du fœtus [ 99 , 100 ]. Les conflits entre gènes imprimés seront donc particulièrement importants pour déterminer les causes de variation de la santé physique, car de nombreux résultats en matière de santé humaine dépendent fortement d’événements survenant au début de la vie et de phénotypes tels que la fonction placentaire et le poids à la naissance [ 101 ], ainsi que du rôle des cellules souches dans le renouvellement et la réparation des tissus.
Prenons par exemple le conflit intragénomique entre les gènes imprimés exprimés par la mère et par le père chez un fœtus, qui visent à prélever des ressources de la mère. Les gènes hérités de la mère chez le fœtus sont sélectionnés pour extraire moins de ressources de la mère que les gènes hérités du père, en raison de leur histoire évolutive de parenté plus élevée avec la parenté matrilinéaire sous un certain degré de paternité multiple. Ce conflit a deux effets. Tout d'abord, au sein d'un locus, il entraîne le silençage du gène codant pour un niveau d'expression inférieur (c'est-à-dire la copie héritée de la mère si le gène code pour l'extraction de ressources maternelles, et la copie héritée du père si le gène code pour la préservation des ressources maternelles) [ 102 ]. Ensuite, entre les locus, le conflit entraîne une escalade de l'expression des locus codant pour des phénotypes opposés.
Pour le phénotype spécifique « pression artérielle maternelle » au milieu ou à la fin de la grossesse, une valeur plus élevée se traduit par une plus grande quantité de nutriments atteignant le fœtus. On s'attend donc à ce qu'un locus codant pour une pression artérielle plus élevée soit exprimé uniquement par le père, en raison d'un conflit intralocus. Un locus codant pour le phénotype opposé, une pression artérielle plus basse, devrait être exprimé par la mère. Les deux locus seront sélectionnés pour augmenter leurs niveaux d'expression en raison d'un conflit interlocus [ 103 ]. Une mutation de perte de fonction dans un allèle à un locus sans conflit devrait avoir de faibles effets phénotypiques, car l'autre allèle serait exprimé. Cependant, dans un locus de conflit, les effets seraient doubles : (i) comme le locus est fonctionnellement haploïde, le produit génique serait absent lorsque la mutation est héritée par la mère, et (ii) comme il existe un équilibre dynamique de tiraillement entraîné par la tension entre les locus d'hypertension et d'hypotension artérielle, une réduction de la production de produits géniques favorisant l'hypotension artérielle devrait entraîner une pathologie à grande échelle, pouvant conduire ici potentiellement à la prééclampsie. Un ensemble de gènes liés à la prééclampsie sont en effet imprimés [ 104 ], et une tension artérielle maternelle modérément mais pas trop élevée entraîne un poids de naissance supérieur à la normale [ 105 ].
Contrairement aux situations impliquant la mère et le fœtus, la modélisation mathématique suggère que les gènes hérités de la mère exprimés à l'âge adulte peuvent dans certaines circonstances être moins liés aux nourrissons et aux jeunes de leur groupe social que les gènes hérités du père [ 106 , 107 ]. Les gènes hérités du père chez les adultes peuvent ainsi être sélectionnés pour promouvoir plus d'altruisme (par exemple, soins parentaux communautaires) envers les nourrissons et les jeunes de leur groupe social que les gènes hérités de la mère. Pour le phénotype « fertilité féminine », cette situation se traduit par une production plus élevée de sa propre progéniture et donc moins de soins communautaires par rapport à la production d'un nombre moins élevé de progénitures et donc plus de soins communautaires.
On peut prédire qu'un locus codant pour une fertilité plus élevée sera réduit au silence par le père, en raison d'un conflit intragénomique. Cependant, on s'attend à ce qu'un locus codant pour le phénotype opposé, une fertilité plus faible, soit réduit au silence par la mère [ 108 ]. Les deux locus seront sélectionnés pour augmenter leurs niveaux d'expression en raison d'un conflit interlocus. On s'attend à ce qu'une mutation avec perte de fonction sur un locus codant pour la fertilité ait le double effet habituel dans les locus conflictuels : la perte de ce produit génique particulier lorsqu'il est hérité de la mère en raison d'une haploïdie fonctionnelle, et une cascade de pathologies liées à une faible fertilité (y compris une insuffisance ovarienne prématurée) en raison de déséquilibres entre les gènes codant pour une fertilité plus élevée et plus faible [ 108 ]. Ces exemples illustrent également comment, pour les gènes imprimés, la théorie peut prédire les formes de phénotypes pathologiques attendus en cas de dysrégulation, ce qui permet d'établir des liens étroits entre la théorie et la médecine clinique.
Les gènes imprégnés sont remarquablement peu étudiés du point de vue de l'évolution, malgré le soutien apporté à la théorie de la parenté de l'empreinte en ce qui concerne la croissance et l'importance considérable de ces gènes pour la croissance, le cancer et les maladies métaboliques [ 89 , 109 ]. Le fait que les gènes eux-mêmes présentent un comportement conflictuel et coopératif [ 110 ] devrait motiver les biologistes comportementaux et évolutionnistes à analyser les mécanismes moléculaires par lesquels ils interagissent socialement et, par conséquent, assurent la médiation de la santé et de la maladie.
(e) Les conflits liés aux ressources entre parents, en particulier les conflits mère-enfant, potentialisent et modulent les risques de maladie
La coopération entre individus génétiquement apparentés représente le contexte original de la théorie de l'aptitude inclusive de Hamilton, qui a été développée pour aider à comprendre l'évolution paradoxale de l'altruisme. Trivers [ 111 ] a été le premier à souligner que les proches parents sont aussi généralement des concurrents proches et que de nombreuses interactions parents-enfants et frères et sœurs peuvent être interprétées dans le contexte de conflits plus ou moins restreints. Considérant un phénotype particulier résultant de l'interaction de deux individus, la valeur phénotypique qui maximise l'aptitude inclusive d'un individu peut être différente de la valeur qui maximise l'aptitude inclusive de l'autre individu. Il existe donc une région de l'espace phénotypique où les changements de valeur augmentent l'aptitude inclusive d'un individu aux dépens de l'autre. La valeur phénotypique qui sera finalement exprimée dépend des forces de sélection des deux parties et de l'individu qui a le plus de contrôle sur le phénotype. Ainsi, un individu peut finir par prendre le contrôle de l'expression phénotypique, en raison d'asymétries physiques et physiologiques. De tels conflits sont basés sur une parenté identique avec des interlocuteurs sociaux à travers des gènes autosomiques non imprimés, les agents numériquement prédominants dans les génomes, qui sont censés s'accorder sur les tactiques de comportement social envers leurs proches.
Les conflits entre parents sont pertinents d’un point de vue médical car ils peuvent augmenter les risques de développer des maladies associées à des changements dans la valeur sélective inclusive. De tels changements impliquent généralement le contrôle de ressources vitales pour la croissance et le développement. Par exemple, Haig [ 112 ] a démontré que de nombreux troubles majeurs de la grossesse humaine, notamment le diabète gestationnel, la prééclampsie et le retard de croissance intra-utérin, pourraient être interprétés comme résultant, en partie, de conflits mère-enfant concernant les ressources maternelles pendant la grossesse. Considérant, par exemple, le phénotype « taux de glucose dans le sang maternel », le fœtus est sélectionné pour maintenir des niveaux de glucose plus élevés (par la libération placentaire de substances chimiques dans le sang maternel qui augmentent sa résistance à l’insuline) que ceux que la mère est sélectionnée pour maintenir. La mère produit donc plus d’insuline. Des séquelles sur la santé surviennent si ce système évolué de tiraillement devient dérégulé, ou si le fœtus « gagne » de telle sorte que la mère développe un diabète gestationnel. Des conflits comparables entre parents sont attendus chaque fois qu’ils sont en compétition pour des ressources liées à la fitness, les résultats des conflits influençant la vulnérabilité et les phénotypes de la maladie.
(f) Les interactions sociales avec les proches dès le début de la vie génèrent et exacerbent les risques de troubles mentaux
Les conflits mère-enfant peuvent être d’ordre psychologique ou physiologique. La parturition représente un point de transition entre les conflits liés à l’investissement des ressources physiques, avec un avantage pour la progéniture à certains égards, et les conflits liés aux interactions comportementales et psychologiques, avec un contrôle maternel prédominant sur la plupart des activités. Comme le placenta et le sein fournissent des ressources énergétiques à la progéniture en développement, les interactions psychologiques, en particulier avec la mère, fournissent des ressources cognitives et émotionnelles qui soutiennent le développement psychologique de la petite enfance, avec des répercussions à vie sur la santé psychologique et le bien-être.
Le concept d'attachement de John Bowlby, à savoir le lien psychologique de l'enfant qui favorise un développement optimal de la cognition sociale et des émotions, sert de construction et de mesure primaires pour comprendre les interactions mère-enfant et leurs conséquences [ 113 , 114 ]. L'attachement de l'enfant peut être (i) sécure, de sorte que l'enfant développe une attente continue d'investissement de la part de la mère, ou (ii) insécure, soit anxieux-insécurisant, dans lequel les enfants souffrant de sollicitations de besoins non satisfaits expriment une détresse et une recherche de contact accrues, combinées à de la colère et de l'ambivalence, soit (iii) évitant-insécurisant, dans lequel les enfants ayant des attentes insatisfaites en viennent à éviter et à rejeter les personnes qui s'occupent d'eux [ 115 ]. La première catégorie peut être interprétée comme des tentatives actives d'améliorer une situation sous-optimale pour l'enfant, tandis que la seconde peut représenter un comportement conçu pour éviter d'aggraver une mauvaise situation [ 114 , 116 ]. Malgré le fait qu’un tel attachement insécurisant soit fortement associé à un large éventail de troubles psychologiques et psychiatriques [ 117 , 118 ], son incidence est remarquablement élevée, de l’ordre d’un tiers des enfants [ 119 ].
Comment ces considérations impliquent-elles la théorie de l’aptitude inclusive et leur impact sur la santé ? La théorie de l’attachement, bien que fondée explicitement sur l’intégration des principes évolutionnistes et éthologiques, n’a pas encore intégré les effets conflictuels de l’aptitude inclusive dans ses préceptes fondamentaux. L’attachement implique essentiellement la sollicitation psychologique de l’enfant et la réponse (ou non) de la personne qui s’en occupe, généralement la mère. Comme décrit ci-dessus, les gènes autosomiques de la progéniture ont été soumis à une sélection pour solliciter plus d’investissement physique de la part des mères que ce qu’ils ont été sélectionnés pour fournir, et les gènes imprimés exprimés par le père favorisent les phénotypes qui augmentent également la sollicitation tandis que les gènes imprimés exprimés par la mère favorisent l’inverse. Ces considérations s’appliquent également à l’investissement comportemental et psychologique, en particulier chez les humains pour qui le développement précoce du cerveau social revêt une importance primordiale pour la santé mentale. Pour toute dyade mère-progéniture donnée, la santé psychologique de l’enfant dans le contexte de l’attachement devrait donc être une fonction de la correspondance entre la sollicitation de l’enfant et l’offre maternelle, et les niveaux d’investissement maternel qui en résultent combinés à son efficacité [ 114 ].
Ce système d’offre et de demande présente deux caractéristiques clés, du point de vue de l’inclusion. Premièrement, en supposant un contrôle maternel prédominant sur les phénotypes liés à l’attachement et des compromis affectant l’allocation maternelle des ressources dans ce contexte, l’attachement insécurisé est alors attendu dans une proportion substantielle de cas, comme documenté ci-dessus. Cette asymétrie est une simple conséquence du contrôle maternel, qui permet aux mères d’atteindre leur niveau optimal d’investissement lié à l’attachement. Deuxièmement, le développement des interactions d’attachement peut être perturbé, par des facteurs génétiques, épigénétiques et environnementaux médiatisés par des systèmes de conflit évolués, dans l’une des deux directions opposées : (i) vers un attachement insécurisé en raison d’une sous-sollicitation, d’une sous-fourniture ou des deux, ou (ii) vers un attachement « trop sûr » en raison d’une combinaison de sur-sollicitation et de sur-fourniture. De plus, les mécanismes immédiats devraient impliquer des effets conflictuels d’inclusion, et la direction et l’ampleur de ces perturbations devraient être directement et causalement liées à des résultats spécifiques en matière de santé psychologique et psychiatrique. Pour un enfant, les biais paternels dans l'expression des gènes imprimés dans le cerveau sont donc associés à une sollicitation excessive ; en revanche, les biais maternels devraient être associés à une sollicitation réduite [ 114 ]. Dans quelle mesure ces attentes sont-elles satisfaites ?
Les systèmes d'attachement ont évolué à l'origine chez les mammifères placentaires souches, avec l'avènement de la viviparité, des soins maternels intensifs, de la lactation, des cerveaux relativement gros et de l'empreinte génomique [ 83 ]. Le neuropeptide ocytocine, qui est également apparu à cette époque, sert de mécanisme moléculaire primaire pour le lien mère-progéniture, dans les deux sens, en plus de ses rôles dans le déclenchement du travail, la lactation et la succion du nourrisson [ 120 ]. Des preuves provenant de souris et d'humains indiquent que l'inactivation de l'un des gènes imprimés exprimés par le père conduit à des réductions substantielles du nombre de neurones producteurs d'ocytocine dans l'hypothalamus et à des altérations de la succion chez les nouveau-nés [ 120 – 124 ]. De plus, deux des gènes impliqués, PEG3 et NDN , représentent des pôles centraux d'un réseau de coexpression de gènes imprimés décrit par Varrault et al . [ 125 ]. Ces résultats indiquent que l’expression des gènes soumis à l’empreinte exerce des effets importants sur le développement du système ocytocinétique dans le cerveau, ce qui a un impact sur les coûts imposés par la progéniture aux mères dans le sens prédit par la théorie de l’empreinte de parenté. Ainsi, les conflits intragénomiques sont également impliqués dans les phénotypes psychologiques médiés par l’ocytocine chez l’homme, qui comprennent la prosocialité, l’empathie, la confiance, les conceptualisations intragroupe-exogroupe, la perception de la parenté et l’altruisme [ 126 – 131 ].
Ces considérations suggèrent que l'ocytocine peut servir de médiateur hormonal clé pour maximiser la capacité d'inclusion, de sorte que des degrés plus proches de parenté ou la perception de la saillance et des avantages de la capacité d'inclusion suscitent une libération et une réponse d'ocytocine plus fortes. Ainsi, les altérations du système ocytocinergique (et des systèmes dopaminergique, sérotoninergique et autres systèmes endocriniens en interaction) devraient avoir des effets disproportionnés sur la santé psychologique, interprétés dans le contexte de la capacité à maximiser inconsciemment sa capacité d'inclusion. Des preuves directes des effets de l'ocytocine et des effets d'empreinte sur l'attachement et ses phénotypes psychiatriques et psychologiques associés chez l'homme proviennent d'études sur trois troubles neurogénétiques impliquant une dysrégulation des gènes imprimés.
Premièrement, le syndrome de Prader-Willi est causé par la perte de l'expression d'un gène paternel imprimé pour un ensemble de gènes du chromosome 15 (provoquant un biais maternel dans le développement) ; il est caractérisé dans la petite enfance par de faibles niveaux de sollicitation exprimés par des pleurs réduits et faibles, un sommeil prolongé, une succion réduite et un faible attachement à la mère [ 93 , 132 ]. Le nombre de neurones à ocytocine dans l'hypothalamus et l'ocytocine sérique sont considérablement réduits dans cette condition [ 133 ]. À l'âge adulte, une forte proportion de personnes atteintes du syndrome de Prader-Willi développent également des formes de schizophrénie ou de psychose affective (psychose qui comprend la dépression), ce qui en fait l'une des causes les plus pénétrantes des troubles psychotiques-affectifs [ 134 ] ; de plus, l'attachement insécurisé dans l'enfance est fortement associé à d'autres troubles psychotiques-affectifs, dont la schizophrénie [ 117 ]. Les liens de cause à effet entre l’attachement insécurisant et les troubles psychotiques-affectifs nécessitent des études plus approfondies, mais ils peuvent inclure un sous-développement du « cerveau paternel » limbique centré sur l’hypothalamus par rapport au « cerveau maternel » mentaliste néocortical [ 135 ], ainsi que la négligence, les mauvais traitements et les abus pendant l’enfance qui dérégulent le développement cognitivo-affectif [ 114 , 117 , 118 ].
Deuxièmement, le syndrome d'Angelman est causé par la perte de fonction du gène UBE3A exprimé par la mère, ce qui produit ainsi un biais en faveur des intérêts du gène paternel imprimé dans le développement [ 135 ]. Pendant la petite enfance, les individus atteints du syndrome d'Angelman montrent des degrés excessifs de sollicitation envers leurs parents, comme l'impliquent des niveaux extrêmes de pleurs, d'hyperactivité et d'insomnie ; ils montrent cependant également des niveaux élevés d'humeur positive, notamment dans les interactions sociales [ 136 , 137 ]. Pris ensemble, ces traits ont été interprétés comme essentiellement l'opposé psychologique du syndrome de Prader-Willi, par lequel l'influence accrue du gène paternel imprimé favorise une sollicitation accrue via l'activité et l'humeur positive. Les niveaux de sérotonine et de dopamine sont élevés dans plusieurs régions du cerveau des souris UBE3A knockout [ 138 ], mais l'ocytocine n'a pas été étudiée ; de même, l'attachement à la mère n'a pas encore été étudié.
Français La plupart des enfants atteints du syndrome d'Angelman et des troubles fortement superposés que sont le syndrome de Rett et le syndrome de Pitt-Hopkins présentent des troubles du spectre autistique [ 139 ], avec un développement réduit des compétences mentales, néocorticales et sociocognitives. Étonnamment, parmi les individus atteints d'un autisme de haut niveau, la fréquence de l'attachement insécurisant n'est pas augmentée au-delà des niveaux normatifs [ 140 ]. Les enfants autistes ont certainement tendance à imposer des exigences de temps et d'énergie plus élevées aux parents, en particulier à la mère, mais le degré auquel les phénotypes du spectre autistique moins extrêmes et non pathologiques impliquent un attachement « trop sûr », qui conserve des éléments de la petite enfance, une dépendance psychologique simple à la mère [ 141 ], reste à évaluer.
Troisièmement, le syndrome de Williams est causé par une petite délétion chromosomique qui comprend le facteur de transcription GTF2I, un gène qui présente un fort biais d'expression spécifique aux parents indiquant une empreinte génomique [ 142 ]. Ce syndrome implique un comportement hypersocial, dans lequel les enfants affectés présentent des niveaux élevés d'approche, d'engagement social et de contact visuel [ 143 , 144 ]. GTF2I a été impliqué dans les altérations sociocomportementales du syndrome de Williams, peut-être par association avec les niveaux environ trois fois plus élevés d'ocytocine trouvés dans cette situation [ 145 ]. Étant donné que GTF2I présente un biais d'expression du gène maternel [ 146 ], une dose réduite de son produit génique (comme dans le syndrome de Williams) génère un biais d'expression paternelle. Le syndrome de Williams est donc similaire au syndrome d'Angelman dans le sens où ses principaux phénotypes comportementaux peuvent être interprétés dans le contexte de comportements exagérés liés à l'attachement, en conjonction avec des biais paternels à l'expression du gène imprimé. Malgré la sollicitation hypersociale observée dans le syndrome de Williams, ce syndrome a également été associé, comme le syndrome d'Angelman, à des compétences sociales réduites et à des phénotypes du spectre autistique [ 146 ]. En revanche, les duplications de la région génomique du syndrome de Williams, dont on peut prédire qu'elles impliquent des niveaux réduits d'ocytocine et un biais génétique maternel, ont récemment été associées à un risque accru de schizophrénie [ 147 ], en parallèle apparent avec le syndrome de Prader-Willi tel que décrit ci-dessus.
Les troubles neurogénétiques tels que le syndrome de Prader-Willi, le syndrome d’Angelman et le syndrome de Williams impliquent des altérations majeures des systèmes évolués de cognition et d’affect (humeur), mais leurs phénotypes et leurs causes génétiques représentent les extrémités de continuums qui devraient évoluer vers la normalité. Ainsi, la variation de l’expression et des effets des gènes imprimés, des composants du système ocytocinergique et d’autres causes de diversité dans les modèles d’attachement de l’enfance et de développement social précoce devraient exercer de fortes répercussions sur le développement de la coopération, de l’altruisme, de l’empathie et de la sensibilité aux altérations de la socialité et du comportement exprimées dans les troubles de la personnalité et les troubles psychiatriques.
En voici trois exemples schématiques :
1/ Le trouble de la personnalité dépendante implique des niveaux extrêmes de comportement lié à la dépendance et à l'attachement en conjonction avec une expression élevée et maladaptée de la dimension de personnalité du modèle à cinq facteurs de l'agréabilité [ 148 ]. Cette condition a été interprétée comme impliquant des niveaux pathologiquement élevés d'altruisme et de sacrifice de soi [ 148 , 149 ], et peut être médiée par un attachement « trop sûr ». En revanche, les troubles de la personnalité narcissique et antisociale peuvent être interprétés dans le contexte de niveaux pathologiquement élevés d'égoïsme (et de faible agréabilité) [ 150 ], à l'extrême opposé. Ces considérations suggèrent la possibilité de « troubles psychologiques de maximisation de la fitness inclusive », impliquant soit l'hyperaltruisme (avec un comportement exprimé même si rb −c < 0) soit l'hypoaltruisme (avec(nécessaire à l’expression comportementale).
2/ Le trouble de la personnalité limite, qui est fortement médiatisé par l'attachement insécurisant et la maltraitance infantile, implique une dysrégulation du système ocytocinergique [ 151 ] mais une amélioration des capacités d'empathie et de « lecture de l'esprit », par rapport aux témoins [ 152 ].
3/ Le névrosisme, l’anxiété et la dépression chez les adolescentes et les jeunes femmes adultes peuvent généralement résulter de niveaux élevés d’empathie et de sensibilité sociale liés à l’altruisme, associés à des environnements familiaux stressants ou abusifs [ 153 , 154 ].
De tels exemples sont importants étant donné la continuité entre la variation normale de la personnalité, les troubles de la personnalité et les troubles psychiatriques [ 155 ], ce qui indique que les considérations issues de la théorie de la fitness inclusive – une clé pour comprendre l’altruisme et la coopération – peuvent fournir des liens solides entre les comportements normaux et dysfonctionnels.
Une dernière considération de la théorie de la fitness inclusive pour la santé psychologique humaine concerne les conflits intragénomiques exprimés dans le cerveau qui affectent la cognition, l'affect et le comportement dans des contextes autres que les interactions mère-enfant [ 156 , 157 ]. Ce sujet, mentionné ci-dessus dans le contexte des syndromes de Prader-Willi, d'Angelman et de Williams, a été largement discuté [ 157 ], en se basant sur l'hypothèse selon laquelle certains troubles psychiatriques, en particulier l'autisme et les troubles psychotiques-affectifs, y compris la schizophrénie, représentent, en partie, des effets extrêmes et inadaptés de biais en faveur de l'expression des gènes imprimés paternels (vers un « cerveau paternel » plus orienté vers soi et moins social), ou maternels (vers un « cerveau maternel » social orienté vers les autres). Empiriquement, cette question peut être abordée en étudiant les effets de la variation génétique et épigénétique des gènes imprimés (et leurs niveaux d'expression) sur les traits psychologiques liés à la socialité et à l'architecture et aux fonctions modulaires du cerveau, en particulier dans les populations non cliniques où la pathologie ne perturbe pas les résultats. Par exemple, le gène imprimé GABRB2 responsable du risque de schizophrénie abrite un allèle SNP qui est associé à la fois à une psychose plus grave dans la schizophrénie et à des degrés plus élevés d'altruisme chez les individus en bonne santé [ 158 ] ; de telles associations d'empreinte avec l'altruisme ont été prédites à partir de modèles développés par Úbeda et Gardner [ 106 , 107 , 159 ]. De même, les SNP du gène imprimé LRRTM1 influencent à la fois le risque de schizophrénie et la latéralité [ 160 ], ce qui suggère que la plus grande échelle de modularité dans le cerveau humain, la différenciation hémisphérique gauche-droite pour le langage et d'autres traits sociaux, est modulée par les effets de l'empreinte et du conflit intragénomique.
L’étude des maladies mentales dans une perspective d’adaptation inclusive nécessite une intégration minutieuse de la psychologie, des neurosciences et de la psychiatrie avec la biologie évolutive. Pour les étudiants en comportement social et en évolution, il est important de comprendre que les troubles psychiatriques peuvent être conceptualisés et étudiés comme des troubles du comportement social généralement adaptatif, de sorte qu’ils peuvent être analysés à l’aide de théories et de méthodes permettant de comprendre la sociabilité humaine et animale typique.
- Discussion
La médecine hamiltonienne représente un domaine central de la médecine évolutionniste car les principaux agents impliqués dans la santé humaine – les microbes, les cellules humaines, les gènes et les individus humains – sont tous sociaux et sont tous régis par les mêmes règles générales et mécanismes visant à maximiser la fitness inclusive. La clé du développement et de l’application de la médecine hamiltonienne devient ainsi la création de liens entre la santé et les causes et conséquences des interactions conflictuelles et coopératives entre parents, y compris l’altruisme, le mutualisme, le parasitisme et la méchanceté. Dans ce contexte, les conflits intragénomiques et intergénomiques médiatisent la santé par l’évolution de systèmes liés aux conflits qui deviennent des cibles de dérégulation, par des écarts par rapport aux optima phénotypiques humains qui découlent de la perpétuation ou de la résolution des conflits, et par des conflits entre allèles ou génotypes de microbes pathogènes, d’autres parasites ou de cellules cancéreuses. En revanche, la coopération peut médiatiser la santé lorsqu’elle se produit entre des agents de maladie génétiquement liés, ou entre d’autres agents liés dont les interactions influencent les risques et les impacts de la maladie.
Dans tous les principes décrits ici, la parenté génétique représente la même variable : la probabilité qu'une entité générant une action sociale soit génétiquement identique par descendance ou transfert horizontal, au locus contrôlant l'action, avec le récepteur, dans le contexte d'une population donnée. Pour les gènes autosomiques chez les humains et d'autres métazoaires, les niveaux de parenté sont des produits directs des liens méiotiques. Cependant, pour les gènes non autosomiques chez les métazoaires, et pour les microbes ou les cellules cancéreuses, les effets de parenté et d'adaptation inclusive doivent être conceptualisés plus soigneusement, en termes d'effets d'un allèle sur la réplication de copies de lui-même (strictement, par rapport aux allèles alternatifs au locus) dans d'autres entités, qu'il s'agisse de groupes de cellules bactériennes pures ou mixtes [ 9 ], de cellules humaines génétiquement plus ou moins divergentes au sein d'un individu [ 79 ], ou d'humains en interaction eux-mêmes. Dans tous ces contextes, la règle d'Hamilton peut également être étendue pour inclure des formes d'interaction sociale basées sur le partage de traits (sélection de « genre ») et la rétroaction des effets des traits sociaux sur la forme physique de l'acteur (sélection de « parentèle ») [ 161 ] ; cette formalisation permet l'application de la règle d'Hamilton à un éventail encore plus large de situations affectant la forme physique et la santé, des gènes aux groupes.
Un deuxième point commun essentiel qui ressort des principes de la médecine hamiltonienne est l'importance du contrôle sur les phénotypes sélectionnés par la parenté qui influencent la santé, car les schémas et les degrés de contrôle influent sur la résolution ou la continuité qui peuvent avoir un impact sur la santé de toutes les parties concernées. Ainsi, une partie peut « gagner » au détriment de la santé de l'autre [ 112 ], une « lutte de pouvoir » dynamique peut s'ensuivre avec des coûts de santé réels ou potentiels pour les deux parties [ 88 ], un conflit peut être réprimé par un agent plus puissant que les parties en compétition (avec des coûts possibles de suppression) [ 162 ] ou les conflits peuvent s'intensifier, avec des coûts croissants et un potentiel d'effets dysrégulateurs [ 91 , 92 , 112 ]. Les résultats de tels conflits ne sont susceptibles d'être prévisibles qu'au cas par cas, car ils dépendent des mécanismes d'interaction proches. De telles considérations devraient obliger à une collaboration étroite entre les scientifiques médicaux et évolutionnistes, et à une formation interdisciplinaire.
Les gènes peuvent être plus ou moins puissants dans les interactions compétitives ou coopératives par le biais d'effets tels qu'une expression plus précoce au cours du développement, la régulation des cellules souches, un degré plus élevé de pléiotropie ou des interactions mutualistes et synergiques avec des gènes qui partagent leurs intérêts en matière d'adaptation inclusive. Les gènes soumis à empreinte semblent représenter des exemples de tels effets, comme décrit ci-dessus, et les gènes du chromosome X peuvent présenter des schémas similaires dans l'expression de l'influence génomique [ 84 , 88 ]. Malgré ces conflits, la grande majorité autosomique des gènes du génome humain suggère que les autosomes et les génomes devraient évoluer vers la répression des conflits intragénomiques liés à la santé, par le biais d'effets tels que la canalisation du développement ou la stabilisation des interactions conflictuelles entre les produits géniques.
Les cellules peuvent également varier en puissance, comme dans le cas des microbes ou des tumeurs, par le biais de signaux adressés à d’autres cellules qui modulent leur expression génétique, leur survie, leur réplication et leurs schémas de différenciation, ou par la sécrétion de produits qui modifient directement le comportement cellulaire. On peut citer ici comme exemples les diverses formes d’interaction sociale microbienne décrites ci-dessus, et les cellules cancéreuses qui sécrètent des facteurs de croissance et d’autres composés pour stimuler la division, la différenciation ou la mort cellulaire [ 60 ]. La capacité à accumuler efficacement des ressources par de tels moyens et à présenter une expansion clonale relativement rapide devrait représenter les cibles de sélection les plus courantes pour les microbes et les cellules cancéreuses, bien que d’autres situations évolutives et écologiques semblent favoriser les cellules qui peuvent persister dans des conditions défavorables ou difficiles, comme le traitement par des agents thérapeutiques.
Les individus et les groupes humains diffèrent évidemment en termes de pouvoir, qu'il soit physiologique (comme entre la mère et le fœtus), physique (comme entre des parties qui diffèrent en force ou en mobilité), social (par le biais de coalitions, d'informations ou d'influences), institutionnel (établi collectivement) ou militaire (conflit) [ 163 ]. Chez les humains, les effets socio-comportementaux sur la santé ne dépendent donc pas seulement des divergences entre individus ou groupes apparentés dans les phénotypes optimaux maximisant la forme physique inclusive, mais aussi des asymétries dans la capacité à atteindre ses optima.
Un troisième aspect remarquable de la médecine hamiltonienne est l’importance qu’elle accorde aux liens de parenté avec les descendants et non descendants, ce qui ajoute une nouvelle dimension à la santé car elle élargit le champ des comportements altruistes qui, par définition, imposent aux acteurs des coûts phénotypiques, potentiellement liés à la santé. Cette considération met en évidence le chevauchement et les divergences entre la santé optimale (par laquelle un état phénotypique est maximisé) et la maximisation de la fitness inclusive (par laquelle les allèles sont sélectionnés en provoquant des états phénotypiques qui augmentent leur succès de réplication). On s’attend donc à ce qu’une bonne santé conduise à une fitness inclusive maximale dans la mesure où elle permet de rechercher les moyens les plus efficaces pour y parvenir. Cependant, étant donné la sélection naturelle de la sénescence, on s’attend de plus en plus à des écarts par rapport à un état de santé optimal à mesure que la valeur reproductive résiduelle diminue après le début de l’âge adulte. Une santé réduite peut, bien sûr, aussi être le résultat direct de la maximisation de la fitness inclusive par le biais d’investissements dans la progéniture et d’autres parents. La fitness inclusive et la santé peuvent donc parfois être incompatibles, mais on attend des individus qu’ils s’efforcent néanmoins d’atteindre une fitness inclusive maximale.
Quelle est la santé mentale optimale, du point de vue de la fitness inclusive ? On prédit que la sélection naturelle ne maximise pas le bonheur ou le bien-être psychologique [ 164 ], mais les efforts continus pour augmenter la fitness inclusive par le biais d’aspects de la cognition, de l’affect et du comportement, en fonction des capacités et de l’environnement de chaque individu. De ce point de vue, les écarts par rapport au bonheur ne sont pas inattendus et devraient prendre des formes particulières et prévisibles. Par exemple, une dépression légère peut être interprétée comme une réponse psychologique à des obstacles perçus ou réels qui entravent la réussite des efforts [ 165 – 167 ], tandis que la manie dans le trouble bipolaire peut être interprétée comme une tentative risquée et incontrôlable [ 168 ]. Dans les deux cas, les troubles psychiatriques représentent des expressions extrêmes et inadaptées de mécanismes de maximisation de la fitness inclusive qui sont bénéfiques dans des formes relativement légères, bien qu’antithétiques au moins au bien-être mental à court terme.
La principale implication de cette perspective est que les traitements de santé mentale qui facilitent le cheminement optimal d'un individu vers une aptitude inclusive maximale devraient être plus efficaces que les traitements qui tentent d'accroître le bonheur ou le bien-être par tout autre moyen. Plus généralement, la santé psychologique peut être considérée en termes de succès continu dans l'effort d'aptitude inclusive, qui ne devrait engendrer des écarts par rapport au bonheur que dans la mesure où la motivation est compensée par le contentement. La distinction fondamentale entre les systèmes neurologiques de « vouloir » par rapport à « aimer » [ 169 ], et leurs associations avec le fonctionnement intégré des systèmes d'ocytocine, de dopamine et de sérotonine, s'accordent directement avec cette perspective d'aptitude inclusive et offrent des pistes claires pour synthétiser les approches immédiates et lointaines.
Quels enseignements les principes de la médecine hamiltonienne nous ont-ils apportés dans cet article, et quelles études médicales et évolutionnistes devraient-ils fortement motiver ? La médecine hamiltonienne a été jusqu’à présent le plus développée dans le contexte des interactions sociales entre les agents pathogènes infectieux humains, en particulier les bactéries. Cependant, la plupart des principaux agents pathogènes humains restent pratiquement non étudiés du point de vue de l’adaptation inclusive, en ce qui concerne (i) la parenté génétique entre les cellules microbiennes en interaction, (ii) la diversité des comportements microbiens coopératifs, compétitifs et socialement exploiteurs affichés par les taxons microbiens (et leurs effets sur la virulence) et (iii) la façon dont les symptômes et les mécanismes de transmission des maladies infectieuses sont modulés par les interactions humaines au sein et entre les familles et les groupes sociaux plus larges. En outre, malgré l’importance croissante des symbiotes microbiens humains mutualistes pour le maintien de la santé humaine, aucun ne semble avoir été étudié, jusqu’à présent, du point de vue de l’adaptation inclusive.
Les approches évolutionnistes et écologiques visant à comprendre et à combattre le cancer se sont multipliées au cours des dix dernières années. Cependant, malgré les études empiriques menées par Gloria Heppner dans les années 1980 et 1990 qui décrivaient les « sociétés de cellules tumorales » comme étant essentielles à la progression du cancer [ 56 ], aucun de ces travaux ne s’appuyait sur des principes de base et établis d’adaptation inclusive pour comprendre et analyser les interactions entre entités plus ou moins apparentées. Nous avons esquissé un cadre pour de telles études, en nous appuyant sur les nombreux parallèles socio-comportementaux, mais aussi sur les différences, entre les cellules microbiennes et les cellules cancéreuses.
Les interactions sociales humaines, en particulier entre proches au début de la vie, semblent représenter l’un des déterminants les plus puissants et les plus répandus de la santé mentale tout au long de la vie, à la fois directement et par le biais des interactions gène-environnement. Nous avons cherché à intégrer les considérations d’aptitude inclusive dans la théorie de l’attachement, le principal cadre théorique pour comprendre les interactions socio-comportementales précoces qui façonnent le développement du cerveau et les risques de troubles de la personnalité et psychiatriques tout au long de la vie. Ce faisant, un rôle potentiel du neuropeptide ocytocine est apparu comme une mesure ou un indicateur de la saillance de l’aptitude inclusive socialement coopérative et altruiste ou mutualiste. L’ocytocine génère-t-elle les plaisirs de la coopération et de la solidarité sociale, en particulier avec les proches, tandis que d’autres hormones telles que la vasopressine et la testostérone servent davantage de mécanismes pour moduler les conceptions cognitives et émotionnelles des coûts et des avantages comportementaux ? Comment les conflits génomiques impliquant le système ocytocinergique modulent-ils la santé psychologique humaine ?
WD Hamilton a adopté une vision sceptique de la médecine moderne qui, selon lui, pourrait, à long terme, conduire à une dépendance excessive à la technologie [ 170 ]. Nous suggérons que la médecine hamiltonienne elle-même, qui se concentre sur les mécanismes évolués pour maximiser la santé humaine en se concentrant sur les comportements sociaux liés à la parenté des cellules, des gènes et des individus, représente une voie utile à suivre – une voie que nous pensons que Hamilton aurait appréciée. Cette approche sera exigeante car elle nécessite que les biologistes à l’esprit évolutionniste, écologique et comportemental maîtrisent des sous-domaines difficiles de la médecine. De telles études auront cependant l’avantage supplémentaire d’accélérer les connaissances sur les principales questions non résolues de la biologie évolutionniste, de l’écologie et du comportement, étant donné que les microbes et les humains représentent également nos systèmes de recherche les mieux compris d’un point de vue mécaniste et immédiat.
First principles of Hamiltonian medicine
Philos Trans R Soc Lond B Biol Sci. 2014 Mar 31;369(1642):20130366
DOI : 10.1098/rstb.2013.0366
Traduction de Luc Perino
_______________________________________________________________________________________________________
Références
1
Williams GW& Nesse RM. 1991 L’aube de la médecine darwinienne. Q. Révérend Biol. 66, 1 à 22. (doi :10.1086/417048).
2
Organisation mondiale de la santé. 2006 Documents de base, 45e éd. Genève, Suisse : OMS.
3
Hamilton WD. 1964 L’évolution génétique du comportement social, I et II. J. Theor. Biol. 7, 1 à 52. (doi :10.1016/0022-5193(64)90038-4).
4
Grafen, A., 2006 Optimisation de la forme physique inclusive. J. Theor. Biol. 238, 541 à 563. (doi :10.1016/j.jtbi.2005.06.009).
5
Foster KR. 2005 Médecine hamiltonienne : pourquoi la vie sociale des agents pathogènes est importante. Science 308, 1269-1270. (doi :10.1126/science.1108158).
6
West SA& Gardner A. 2013 Adaptation et fitness inclusif. Curr. Biol. 23, R577 à R584. (doi :10.1016/j.cub.2013.05.031).
7
Crespi BJ. 2001 L’évolution du comportement social chez les micro-organismes. Tendances Ecol. Evol. 16, 178 à 183. (doi :10.1016/S0169-5347(01)02115-2).
8
West SA, Diggle SP, Buckling A, Gardner A& Griffin AS. 2007 La vie sociale des microbes. Annu. Rev. Ecol. Evol. Syst. 38, 53 à 77. (doi :10.1146/annurev.ecolsys.38.091206.095740).
9
Strassmann JE, Gilbert OM & Queller DC. 2011 Discrimination de la parenté et coopération chez les microbes. Annu. Rev. Microbiol. 65, 349 à 367. (doi :10.1146/annurev.micro.112408.134109).
10
Gardner A, West SA& Buckling A. 2004 Bactériocines, dépit et virulence. Proc. R. Soc. Lond. B 271, 1529 à 1535. (doi :10.1098/rspb.2004.2756).
11
Cotter, Ross RP& Hill C. 2013 Les bactériocines : une alternative viable aux antibiotiques ? Nat. Rev. Microbiol. 11, 95 à 105. (doi :10.1038/nrmicro2937).
12
Ratcliff WC, Hoverman M, Travisano M& Denison RF. 2013 Sélection de parenté cryptique dans l’évolution de la dormance microbienne. Am. Nat. 182, 147 à 156. (doi :10.1086/670943).
13
Reece SE, Drew, DR& Gardner A., 2008, Ajustement du rapport de masculinité et discrimination de la parenté chez les parasites du paludisme. Nature 453, 609 à 614. (doi :10.1038/nature06954).
14
Nadell CD, Xavier JB& Foster KR. 2009 La sociobiologie des biofilms. FEMS Microbiol. Rev. 33, 206 à 224. (doi :10.1111/j.1574-6976.2008.00150.x).
15
Boyle KE, Heilmann S, van Ditmarsch D& Xavier JB. 2013 Exploiter l’évolution sociale dans les biofilms. Curr. Opin. Microbiol. 16, 207 à 212. (doi :10.1016/j.mib.2013.01.003).
16
Smith, J., 2001 L’évolution sociale de la pathogenèse bactérienne. Proc. R. Soc. Lond. B 268, 61 à 69. (doi :10.1098/rspb.2000.1330).
17
Nogueira T, Rankin DJ, Touchon M, Taddei F, Brown SP& Rocha EP. 2009 Le transfert horizontal de gènes du sécrétome est à l’origine de l’évolution de la coopération bactérienne et de la virulence. Curr. Biol. 19, 1683-1691. (doi :10.1016/j.cub.2009.08.056).
18
McGinty SÉ, Lehmann L, Brown SP et Rankin DJ. 2013 L’interaction entre la parenté et le transfert horizontal de gènes est à l’origine de l’évolution des biens publics transportés par les plasmides. Proc. R. Soc. B 280, 20130400. (doi :10.1098/rspb.2013.0400).
19
Jiricny N, Diggle SP, West SA, Evans BA, Ballantyne G, Ross-Gillespie A& Griffin AS. 2010 La valeur adaptative est corrélée à l’ampleur de la tricherie chez une bactérie. J. Evol. Biol. 23, 738 à 747. (doi :10.1111/j.1420-9101.2010.01939.x).
20
Mellbye B& Schuster M. 2011 La sociomicrobiologie de la résistance aux médicaments antivirulents : une preuve de concept. mBio 2, e00131. (doi :10.1128/mBio.00131-11).
21
Poivre JW. 2012 Les médicaments qui ciblent les biens publics pathogènes sont robustes contre l’évolution de la résistance aux médicaments. Evol. Appl. 5, 757 à 761. (doi :10.1111/j.1752-4571.2012.00254.x).
22
Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M& Hardt WD. 2013 Stabilisation de la virulence coopérative par l’expression d’un phénotype avirulent. Nature 494, 353 à 356. (doi :10.1038/nature11913).
23
Tanouchi Y, Pai A, Buchler NE& You L. 2012 Programmation de la mort altruiste induite par le stress chez les bactéries modifiées. Mol. Syst. Biol. 8, 626. (doi :10.1038/msb.2012.57).
24
Berngruber TW, Lion S& Gandon S. 2013 Évolution du suicide comme stratégie de défense contre les agents pathogènes dans un environnement spatialement structuré. Ecol. Lett. 16, 446 à 453. (doi :10.1111/ele.12064).
25
Refardt D, Bergmiller T& Kümmerli R. 2013 L’altruisme peut évoluer lorsque la parenté est faible : preuves du suicide des bactéries lors d’une infection par phage. Proc. R. Soc. B 280, 20123035. (doi :10.1098/rspb.2012.3035).
26
Hentzer M, Eberl L, Nielsen J& Givskov M. 2003 Détection du quorum : une nouvelle cible pour le traitement des infections à biofilm. Biomédicaments 17, 241-250. (doi :10.2165/00063030-200317040-00003).
27
André J-B & Bernard G. 2005 L’organisation multicellulaire chez les bactéries comme cible pour la thérapie médicamenteuse. Ecol. Lett. 8, 800 à 810. (doi :10.1111/j.1461-0248.2005.00783.x). ,
28
Dong Y-H, Wang L-Y et Zhang L-H. 2007 Extinction du quorum : mécanismes et implications. Phil. Trans. R. Soc. B 362, 1201–1211. (doi :10.1098/rstb.2007.2045).
29
Swem LR, Swem DL, O’Loughlin CT, Gatmaitan R, Zhao B, Ulrich SM et Bassler BL. 2009 Un antagoniste sensible au quorum cible à la fois les récepteurs membranaires et cytoplasmiques et contrôle la pathogénicité bactérienne. Mol. Cellule. 35, 143 à 153. (doi :10.1016/j.molcel.2009.05.029).
30
Dunny GM, Brickman TJ & Dworkin M. 2008 Comportement multicellulaire chez les bactéries : communication, coopération, compétition et tricherie. Bioessais 30, 296-298. (doi :10.1002/bies.20740).
31
Travisano M& Velicer GJ. 2004 Stratégies de contrôle des tricheurs microbiens. Tendances Microbiol. 12, 72 à 78. (doi :10.1016/j.tim.2003.12.009).
32
Frank SA. 1992 Un modèle de sélection de parenté pour l’évolution de la virulence. Proc. R. Soc. Lond. B 250, 195 à 197. (doi :10.1098/rspb.1992.0149).
33
Frank SA. 1996 Modèles de virulence du parasite. Q. Révérend Biol. 71, 37 et 78. (doi :10.1086/419267).
34
Lively CM. 2009 Compétition d’hôtes locaux dans l’évolution de la virulence. J. Evol. Biol. 22, 1268 à 1274. (doi :10.1111/j.1420-9101.2009.01743.x).
35
Buckling A& Brockhurst MA. 2008 La sélection de la parenté et l’évolution de la virulence. L’hérédité (Edinb.) 100, 484 à 488. (doi :10.1038/sj.hdy.6801093).
36
Ross-Gillespie A, Gardner A, West SA et Griffin AS. 2007 Dépendance aux fréquences et coopération : théorie et test avec des bactéries. Am. Nat. 170, 331 à 342. (doi :10.1086/519860).
37
Eldar, A., 2011, Le conflit social est à l’origine de la divergence évolutive de la perception du quorum. Proc. Natl Acad. Sci. États-Unis 108, 13 635–13 640. (doi :10.1073/pnas.1102923108). ,
38
Rumbaugh KP, Trivedi U, Watters C, Burton-Chellew MN, Diggle SP& West SA. 2012 Sélection de la parenté, détection du quorum et virulence chez les bactéries pathogènes. Proc. R. Soc. B 279, 3584 à 3588. (doi :10.1098/rspb.2012.0843).
39
Lion S. 2013 Infections multiples, sélection de la parenté et épidémiologie évolutive des traits parasitaires. J. Evol. Biol. 26, 2107 à 2122. (doi :10.1111/jeb.12207).
40
Mitri, S& Foster, KR., 2013, La vision génotypique des interactions sociales dans les communautés microbiennes. Annu. Révérend Genet. 47, 247 à 273. (doi :10.1146/annurev-genet-111212-133307).
41
Smith J, Van Dyken JD et Zee PC. 2010 Une généralisation de la règle de Hamilton pour l’évolution de la coopération microbienne. Science 328, 1700-1703. (doi :10.1126/science.1189675).
42
Griffin AS, West SA& Buckling A. 2004 Coopération et compétition dans les bactéries pathogènes. Nature 430, 1024 à 1027. (doi :10.1038/nature02744).
43
Dethlefsen L, McFall-Ngai M& Relman DA. 2007 Une perspective écologique et évolutive sur le mutualisme et la maladie entre humains et microbes. Nature 449, 811 à 818. (doi :10.1038/nature06245).
44
Brown SP, Cornforth DM& Mideo N. 2012 Évolution de la virulence chez les pathogènes opportunistes : généralisme, plasticité et contrôle. Tendances Microbiol. 20, 336 à 342. (doi :10.1016/j.tim.2012.04.005).
45
Madupu R, Szpakowski S& Nelson KE. 2013 Le microbiome dans la santé humaine et la maladie. Sci. Prog. 96, 153 à 170. (doi :10.3184/003685013X13683759820813). , PubMed,
46
Alizon S, de Roode JC& Michalakis Y. 2013 Infections multiples et évolution de la virulence. Ecol. Lett. 16, 556 à 567. (doi :10.1111/ele.12076).
47
Allen B& Nowak MA. 2013 La coopération et le destin des sociétés microbiennes. PLoS Biol. 11, e1001549. (doi :10.1371/journal.pbio.1001549).
48
Blower TR, Evans TJ, Przybilski R, Fineran PC& Salmond GP. 2012 L’évasion virale d’un système de suicide bactérien par mimétisme moléculaire basé sur l’ARN permet l’altruisme infectieux. PLoS Genet. 8, e1003023. (doi :10.1371/journal.pgen.1003023).
49
Nonacs P& Kapheim KM. 2012 Modélisation de l’évolution de la maladie avec sélection à plusieurs niveaux : le VIH en tant que génome social de quasi-espèce. J. Evol. Med. 1, 235553. (doi :10.4303/jem/235553). ,
50
Cotter, SC& Kilner, RM. 2010 Immunité personnelle versus immunité sociale. Behav. Ecol. 21, 663 à 668. (doi :10.1093/beheco/arq070). ,
51
Débarre F, Lion S, van Baalen M& Gandon S. 2012 Evolution des traits d’histoire de vie de l’hôte dans un système hôte-parasite structuré spatialement. Am. Nat. 179, 52 à 63. (doi :10.1086/663199).
52
Cors F& Hood ME. 2012 L’évolution de la résistance et de la tolérance aux maladies dans des populations spatialement structurées. Ecol. Evol. 2, 1705-1711. (doi :10.1002/ece3.290).
53
Best A, Webb S, White A& Boots M. 2011 Résistance et coévolution de l’hôte dans les populations spatialement structurées. Proc. R. Soc. B 278, 2216 à 2222. (doi :10.1098/rspb.2010.1978).
54
Schliekelman, P., 2007, Sélection de la parenté et évolution de la résistance aux maladies infectieuses. Évolution 61, 1277-1288. (doi :10.1111/j.1558-5646.2007.00122.x).
55
Scott S& Duncan C. 2004 Le retour de la peste noire : le plus grand tueur en série du monde. West Sussex, Royaume-Uni : John Wiley and Sons.
56
Heppner GH. 1993 Sociétés de cellules cancéreuses et progression tumorale. Cellules souches 11, 199 à 203. (doi :10.1002/stem.5530110306).
57
Axelrod R, Axelrod DE& Pienta KJ. 2006 Évolution de la coopération entre les cellules tumorales. Proc. Natl Acad. Sci. États-Unis 103, 13 474–13 479. (doi :10.1073/pnas.0606053103). ,
58
Michor F& Polyak K. 2010 Les origines et les implications de l’hétérogénéité intratumorale. Cancer Prev. Res. (Phila.) 3, 1361–1364. (doi :10.1158/1940-6207.CAPR-10-0234).
59
Wagstaff L, Kolahgar G& Piddini E. 2013 Interactions cellulaires compétitives dans le cancer : un bras de fer cellulaire. Tendances Cell. Biol. 23, 160 à 167. (doi :10.1016/j.tcb.2012.11.002).
60
Nagy JD& Armbruster D. 2012 Evolution de la prolifération incontrôlée et de l’interrupteur angiogénique dans le cancer. Math. Biosci. Eng. 9, 843 à 876. (doi :10.3934/mbe.2012.9.843).
61
Trejo-Becerril C, et al. 2012 Progression du cancer médiée par transfert horizontal de gènes dans un modèle in vivo. PLoS ONE 7, e52754. (doi :10.1371/journal.pone.0052754).
62
Archetti M. 2013 Dynamique évolutive de l’effet Warburg : la glycolyse comme problème d’action collective parmi les cellules cancéreuses. J. Theor. Biol. 341C, 1 à 8.
63
Hickson J, Diane Yamada S, Berger J, Alverdy J, O’Keefe J, Bassler B& Rinker-Schaeffer C. 2009 Interactions sociétales dans les métastases du cancer de l’ovaire : une hypothèse de détection du quorum. Clin. Exp. Metastasis 26, 67-76. (doi :10.1007/s10585-008-9177-z).
64
Ghosh S, Elankumaran S& Puri IK. 2011 Modèle mathématique du rôle de la signalisation intercellulaire dans la coopération intercellulaire au cours de la tumorigenèse. Cell Prolif. 44, 192 à 203. (doi :10.1111/j.1365-2184.2011.00739.x).
65
Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B& Rak J. 2011 Microvésicules en tant que médiateurs de la communication intercellulaire dans le cancer : la science émergente des « débris » cellulaires. Semin. Immunopathol. 33, 455 à 467. (doi :10.1007/s00281-011-0250-3).
66
Nagy JD. 2004 Compétition et sélection naturelle dans un modèle mathématique du cancer. Bull. Math. Biol. 66, 663 à 687. (doi :10.1016/j.bulm.2003.10.001).
67
Nagy JD. 2008 Les hypertumeurs dans le cancer peuvent être causées par la demande tumorale de phosphore. Proc. Appl. Math. Mech. 7, 1121703. (doi :10.1002/pamm.200700761). ,
68
Aktipis CA& Nesse RM. 2013 Fondements évolutifs de la biologie du cancer. Evol. Appl. 6, 144 à 159. (doi :10.1111/eva.12034).
69
Sprouffske K, Aktipis CA, Radich JP, Carroll M, Nedelcu AM et Maley CC. 2013 Une explication évolutive de la présence de cellules non souches cancéreuses dans les néoplasmes. Evol. Appl. 6, 92 à 101. (doi :10.1111/eva.12030).
70
Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F& Ochiya T. 2012 Interactions compétitives des cellules cancéreuses et des cellules normales via des microARN sécrétoires. J. Biol. Chem. 287, 1397 à 1405. (doi :10.1074/jbc. M111.288662).
71
Agur Z, Kogan Y, Levi L, Harrison H, Lamb R, Kirnasovsky OU et Clarke RB. 2010 La perturbation d’un mécanisme de détection du quorum déclenche la tumorigenèse : un modèle discret simple corroboré par des expériences sur des cellules souches de cancer mammaire. Biol. Direct. 5 et 20. (doi :10.1186/1745-6150-5-20).
72
Krepkin, K& Costa, J., 2011 Définir le rôle de la coopération dans la progression tumorale précoce. J. Theor. Biol. 285, 36 à 45. (doi :10.1016/j.jtbi.2011.06.035).
73
Heppner GH. 1989 Sociétés de cellules tumorales. J. Natl. Cancer Inst. 81, 648 à 649. (doi :10.1093/jnci/81.9.648). , PubMed,
74
Ben-Jacob E, Coffey, DS et Levine, H. 2012 Les stratégies de survie bactérienne suggèrent de repenser la coopérativité du cancer. Tendances Microbiol. 20, 403 à 410. (doi :10.1016/j.tim.2012.06.001).
75
Lambert G, Estévez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD et Austin RH. 2011 Une analogie entre l’évolution de la résistance aux médicaments dans les communautés bactériennes et les tissus malins. Nat. Rev. Cancer 11, 375 à 382. (doi :10.1038/nrc3039).
76
Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL & Holmgren L. 2001 Transfert horizontal d’oncogènes par absorption de corps apoptotiques. Proc. Natl Acad. Sci. États-Unis 98, 6407 à 6411. (doi :10.1073/pnas.101129998).
77
Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO et Skog J. 2011 Les microvésicules tumorales contiennent des éléments rétrotransposons et des séquences oncogènes amplifiées. Nat. Commun. 2, 180. (doi :10.1038/ncomms1180).
78
Murchison EP. 2008 Cancers transmissibles par clonage chez les chiens et les diables de Tasmanie. Oncogène 27, S19 à S30. (doi :10.1038/onc.2009.350). , PubMed,
79
Sprouffske K, Merlo LMF, Gerrish PJ, Maley CC et Sniegowski. 2012 Cancer à la lumière de l’évolution expérimentale. Curr. Biol. 22, R762 à R771. (doi :10.1016/j.cub.2012.06.065).
80
Avner BS, Fialho AM& Chakrabarty AM. 2012 Surmonter la résistance aux médicaments dans les cancers et les microorganismes multirésistants aux médicaments : un cadre conceptuel. Bio-ingénierie 3, 262-270. (doi :10.4161/bioe.21130).
81
Frank SA. 2013 Évolution microbienne : la conception réglementaire empêche les proliférations cancéreuses. Curr. Biol. 23, R343 à R346. (doi :10.1016/j.cub.2013.03.046).
82
Dejosez M, Ura H, Brandt VL& Zwaka TP. 2013 Garanties pour la coopération cellulaire dans l’embryogenèse de souris montrées par le criblage de tricheur à l’échelle du génome. Science 341, 1511-1514. (doi :10.1126/science.1241628).
83
Hamilton WD. 1967 Rapports de masculinité extraordinaires. Une théorie du sex-ratio pour la liaison sexuelle et la consanguinité a de nouvelles implications en cytogénétique et en entomologie. Science 156, 477-488. (doi :10.1126/science.156.3774.477).
84
Haig, D., 2006, Politique intragénomique. Cytogenet. Génome Res. 113, 68 à 74. (doi :10.1159/000090816).
85
Jansen, VAA& van Baalen, M., 2006, L’altruisme par la chromodynamique de la barbe. Nature 440, 663 à 666. (doi :10.1038/nature04387).
86
Biernaskie JM, West SA& Gardner A. 2011 Les barbes vertes sont-elles des hors-la-loi intragénomiques ? Évolution 65, 2729 à 2742. (doi :10.1111/j.1558-5646.2011.01355.x).
87
Stencel A& Crespi B. 2013 Qu’est-ce qu’un génome ? Mol. Ecol. 22, 3437 à 3443. (doi :10.1111/mec.12355).
88
Frank SA& Crespi BJ. 2011 Pathologie du conflit évolutif, avec une théorie du conflit du chromosome X contre l’autosome sur des traits sexuellement antagonistes. Proc. Natl Acad. Sci. États-Unis 108, 10 886 à 10 893. (doi :10.1073/pnas.1100921108). ,
89
Crespi BJ. 2010 Les origines et l’évolution du risque de maladie génétique chez l’homme moderne. Ann. N.Y. Acad. Sci. 1206, 80 à 109. (doi :10.1111/j.1749-6632.2010.05707.x).
90
Úbeda F& Wilkins JF. 2008 Gènes imprimés et maladie humaine : une perspective évolutive. Empreinte génomique (éd. & Wilkins JF), pp. 101-113. New York, NY : Springer.
91
Wilkins JF& Úbeda F. 2011 Maladies associées à l’empreinte génomique. Prog. Mol. Biol. Transl. Sci. 101, 401 à 445. (doi :10.1016/B978-0-12-387685-0.00013-5).
92
Wilkins JF. 2011 Empreinte génomique et décanalisation induite par les conflits. Évolution 65, 537 à 553. (doi :10.1111/j.1558-5646.2010.01147.x).
93
Úbeda F. 2008 Évolution de l’empreinte génomique avec les soins biparentaux : implications pour les syndromes de Prader-Willi et Angelman. PLoS Biol. 6, E208. (doi :10.1371/journal.pbio.0060208).
94
Eggermann T, Eggermann K& Schönherr N. 2008 Retard de croissance versus prolifération : le syndrome de Silver-Russell est génétiquement opposé au syndrome de Beckwith-Wiedemann. Tendances Genet. 24, 195 à 204. (doi :10.1016/j.tig.2008.01.003).
95
Soejima H& Higashimoto K., 2013 Altérations épigénétiques et génétiques du syndrome de Beckwith-Wiedemann et des troubles apparentés. J. Hum. Genet. 58, 402 à 409. (doi :10.1038/jhg.2013.51).
96
Andersen DC, Laborda J, Baladron V, Kassem M, Sheikh SP & Jensen CH. 2013 Double rôle de l’homologue delta-like 1 (DLK1) dans le développement des muscles squelettiques et la régénération musculaire adulte. Développement 140, 3743 à 3753. (doi :10.1242/dev.095810).
97
Venkatraman A, et al. 2013 L’empreinte maternelle au locus H19-Igf2 maintient la quiescence des cellules souches hématopoïétiques adultes. Nature 500, 345 à 349. (doi :10.1038/nature12303).
98
Schmidt-Edelkraut U, Hoffmann A, Daniel G& Spengler D. 2013 Zac1 régule la différenciation astrogliale des cellules souches neurales par le biais de socs3. Cellules souches 31, 1621-1632. (doi :10.1002/stem.1405).
99
Renfree MB, Suzuki S& Kaneko-Ishino T. 2013 L’origine et l’évolution de l’empreinte génomique et de la viviparité chez les mammifères. Phil. Trans. R. Soc. B 368, 20120151. (doi :10.1098/rstb.2012.0151).
100
Frost JM et Moore GE. 2010 L’importance de l’empreinte dans le placenta humain. PLoS Genet. 6, e1001015. (doi :10.1371/journal.pgen.1001015).
101
Barker DJ, Lampl M, Roseboom T& Winder N. 2012 Allocation des ressources in utero et santé plus tard dans la vie. Placenta 33, e30–e34. (doi :10.1016/j.placenta.2012.06.009).
102
Úbeda F& Haig D. 2003 Diviser l’enfant. Genetica 117, 103 à 110. (doi :10.1023/A :1022320801661).
103
Wilkins JF& Haig D. 2001 Empreinte génomique de deux loci antagonistes. Proc. R. Soc. Lond. B 268, 1861-1867. (doi :10.1098/rspb.2001.1651).
104
Choudhury M& Friedman JE. 2012 Épigénétique et microARN dans la prééclampsie. Clin. Exp. Hypertens. 34, 334 à 341. (doi :10.3109/10641963.2011.649931).
105
Naeye RL. 1981 Pression artérielle maternelle et croissance fœtale. Am. J. Obstet. Gynecol. 141, 780 à 787.
106
Úbeda F& Gardner A. 2011 Un modèle d’empreinte génomique dans le cerveau social : les adultes. Évolution 65, 462 à 475. (doi :10.1111/j.1558-5646.2010.01115.x).
107
Úbeda F& Gardner A. 2012 Un modèle d’empreinte génomique dans le cerveau social : les aînés. Évolution 66, 1567-1581. (doi :10.1111/j.1558-5646.2011.01517.x).
108
Úbeda F, Ohtsuki H& Gardner A. 2013 L’écologie est à l’origine d’un conflit intra-génomique sur la ménopause. Ecol. Lett. 17, 165 à 174. (doi :10.1111/ele.12208).
109
Crespi, B., 2011 La biologie évolutive de la santé infantile. Proc. R. Soc. B 278, 1441-1449. (doi :10.1098/rspb.2010.2627).
110
Springer SA, Crespi BJ et Swanson WJ. 2011 Au-delà du gambit phénotypique : l’écologie comportementale moléculaire et l’évolution de l’architecture génétique. Mol. Ecol. 20, 2240 à 2257. (doi :10.1111/j.1365-294X.2011.05116.x).
111
Trivers RL. 1974 Conflit parent-progéniture. Am. Zool. 14, 249 à 264. (doi :10.1093/icb/14.1.249). ,
112
Haig, D., 1993 Conflits génétiques dans la grossesse humaine. Q. Révérend Biol. 68, 495 à 532. (doi :10.1086/418300).
113
Bowlby J. 1999 Attachement : attachement et perte, 2e éd., vol. 1. New York, NY : Livres de base.
114
Crespi BJ. 2011 Les stratégies des gènes : conflits génomiques, théorie de l’attachement et développement du cerveau social. Cerveau, comportement et épigénétique (eds, Petronis A& Mill J), pp. 143-168. Heidelberg, Berlin : Springer.
115
Shaver PR& Mikulincer M., 2002 Psychodynamique liée à l’attachement. Attacher. Hum. Dev. 4, 133 à 161. (doi :10.1080/14616730210154171). , PubMed,
116
Chisholm JS. 1996 L’écologie évolutive de l’organisation de l’attachement. Hum. Nat. 7, 1 à 38. PubMed,
117
Benoit D. 2004 L’attachement nourrisson-parent : définition, types, antécédents, mesures et résultats. Paediatr. Santé de l’enfant 9, 541-545. , PubMed,
118
Berry K, Barrowclough C& Wearden A. 2007 Un examen du rôle du style d’attachement adulte dans la psychose : problèmes et questions inexplorés pour des recherches ultérieures. Clin. Psychol. Rev. 27, 458 à 475. (doi :10.1016/j.cpr.2006.09.006).
119
Prior V& Glaser D. 2006 Comprendre l’attachement et les troubles de l’attachement : théorie, preuves et pratique. Londres, Royaume-Uni : Jessica Kingsley Publishers.
120
Schaller F, Watrin F, Sturny R, Massacrier A, Szepetowski P& Muscatelli F. 2010 Une seule injection postnatale d’ocytocine sauve le comportement alimentaire létal chez les nouveau-nés de souris déficients pour le gène Magel2 imprimé. Hum. Mol. Genet. 19, 4895 à 4905. (doi :10.1093/hmg/ddq424).
121
Champagne FA, Curley JP, Swaney WT, Hasen NS et Keverne EB. 2009 Influence paternelle sur le comportement féminin : le rôle de Peg3 dans l’exploration, l’olfaction et la régulation neuroendocrinienne du comportement maternel des souris femelles. Se comporter. Névrose. 123, 469 à 480. (doi :10.1037/a0015060).
122
Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P& Cremer H. 2000 La perturbation du gène Necdin de la souris entraîne des altérations hypothalamiques et comportementales rappelant le syndrome de Prader-Willi chez l’homme. Hum. Mol. Genet. 9, 3101 à 3110. (doi :10.1093/hmg/9.20.3101).
123
Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J& Kelsey G. 2004 La protéine de signalisation imprimée XLαs est nécessaire à l’adaptation postnatale à l’alimentation. Nat. Genet. 36, 818 à 826. (doi :10.1038/ng1397).
124
Curley JP. 2011 Existe-t-il un cerveau social à empreinte génomique ? Bioessais 33, 662-668. (doi :10.1002/bies.201100060).
125
Varrault A, et al. 2006 Zac1 régule un réseau de gènes imprégnés qui joue un rôle critique dans le contrôle de la croissance embryonnaire. Cellule de développement 11, 711 à 722. (doi :10.1016/j.devcel.2006.09.003).
126
Feldman, R., 2012 : Ocytocine et affiliation sociale chez l’homme. Horm. Se comporter. 61, 380 à 391. (doi :10.1016/j.yhbeh.2012.01.008).
127
Veening JG & Olivier B. 2013 Administration intranasale d’ocytocine : effets comportementaux et cliniques, une revue. Névrose. Biobehav. Apocalypse 37, 1445-1465. (doi :10.1016/j.neubiorev.2013.04.012).
128
De Dreu CK, Greer LL, Van Kleef GA, Shalvi S& Handgraaf MJ. 2011 L’ocytocine favorise l’ethnocentrisme humain. Proc. Natl Acad. Sci. États-Unis 108, 1262 à 1266. (doi :10.1073/pnas.1015316108).
129
Fischer-Shofty M, Levkovitz Y& Shamay-Tsoory SG. 2013 L’ocytocine facilite la perception précise de la compétition chez les hommes et de la parenté chez les femmes. Soc. Cogn. Affect. Névrose. 8, 313 à 317. (doi :10.1093/scan/nsr100).
130
Fischer-Shofty M, Brüne M, Ebert A, Shefet D, Levkovitz Y& Shamay-Tsoory SG. 2013 Améliorer la perception sociale dans la schizophrénie : le rôle de l’ocytocine. Schizophr. Res. 146, 357 à 362. (doi :10.1016/j.schres.2013.01.006).
131
Preston SD. 2013 Les origines de l’altruisme dans les soins à la progéniture. Psychol. Bull. 139, 1305 à 1341. (doi :10.1037/a0031755).
132
Haig, D& Wharton, R., 2003 : Le syndrome de Prader-Willi et l’évolution de l’enfance humaine. Am. J. Hum. Biol. 15, 320 à 329. (doi :10.1002/ajhb.10150).
133
Swaab DF, Purba JS et Hofman MA. 1995 Altérations du noyau paraventriculaire hypothalamique et de ses neurones d’ocytocine (cellules de satiété présumées) dans le syndrome de Prader-Willi : une étude de cinq cas. J. Clin. Endocrinol. Metab. 80, 573 à 579. PubMed,
134
Soni S, Whittington J, Holland AJ, Webb T, Maina EN, Boer H& Clarke D. 2008 La phénoménologie et le diagnostic de la maladie psychiatrique chez les personnes atteintes du syndrome de Prader-Willi. Psychol. Med. 38, 1505 à 1514. (doi :10.1017/S0033291707002504).
135
Mabb AM, Judson MC, Zylka MJ & Philpot BD. 2011 Syndrome d’Angelman : aperçu de l’empreinte génomique et des phénotypes neurodéveloppementaux. Tendances Neurosci. 34, 293 à 303. (doi :10.1016/j.tins.2011.04.001).
136
Oliver C, Horsler K, Berg K, Bellamy G, Dick K& Griffiths E. 2007 L’empreinte génomique et l’expression de l’affect dans le syndrome d’Angelman : qu’y a-t-il dans le sourire ? J. Child Psychol. Psychiatry 48, 571-579. (doi :10.1111/j.1469-7610.2007.01736.x).
137
Bernard D. 2008 Syndrome d’Angelman. Hoboken, NJ : Wiley-Blackwell.
138
Farook MF, DeCuypere M, Hyland K, Takumi T, LeDoux MS et Reiter LT. 2012 Niveaux modifiés de sérotonine, de dopamine et de noradrénaline dans des modèles murins de duplication 15q et de syndrome d’Angelman. PLoS ONE 7, e43030. (doi :10.1371/journal.pone.0043030).
139
Jedele KB. 2007 Le spectre chevauchant des syndromes de Rett et d’Angelman : une revue clinique. Semin. Pédiatr. Neurol. 14, 108 à 117. (doi :10.1016/j.spen.2007.07.002). , PubMed,
140
Rutgers AH, Bakermans-Kranenburg MJ, van Ijzendoorn MH et van Berckelaer-Onnes IA. 2004 Autisme et attachement : une revue méta-analytique. J. Child. Psychiatrie psychologique 45, 1123-1134. (doi :10.1111/j.1469-7610.2004.T01-1-00305.x).
141
Crespi B. 2013 Hétérochronie développementale et évolution de la perception, de la cognition et du comportement autistiques. BMC Med. 11 et 119. (doi :10.1186/1741-7015-11-119).
142
Collette JC, Chen XN, Mills DL, Galaburda AM, Reiss AL, Bellugi U& Korenberg JR. 2009 Syndrome de William : l’expression des gènes est liée à l’origine parentale et au contrôle des coordonnées régionales. J. Hum. Genet. 54, 193 à 198. (doi :10.1038/jhg.2009.5).
143
Dilts CV, Morris CA& Leonard CO. 1990 Hypothèse pour le développement d’un phénotype comportemental dans le syndrome de Williams. Am. J. Med. Genet. Suppl. 6, 126 à 131. PubMed,
144
Järvinen A, Korenberg JR& Bellugi U. 2013 Le phénotype social du syndrome de Williams. Curr. Opin. Neurobiol. 23, 414 à 422. (doi :10.1016/j.conb.2012.12.006).
145
Dai L, Carter CS, Ying J, Bellugi U, Pournajafi-Nazarloo H& Korenberg JR. 2012 L’ocytocine et la vasopressine sont dérégulées dans le syndrome de Williams, une maladie génétique affectant le comportement social. PLoS ONE 7, e38513. (doi :10.1371/journal.pone.0038513).
146
Tordjman S, et al. 2012 Trouble autistique chez les patients atteints du syndrome de Williams-Beuren : une reconsidération du phénotype du syndrome de Williams-Beuren. PLoS ONE 7, e30778. (doi :10.1371/journal.pone.0030778).
147
Mulle JG, et al. 2013 La duplication réciproque de la délétion du syndrome de Williams-Beuren sur le chromosome 7q11.23 est associée à la schizophrénie. Biol. Psychiatrie. 75, 371 à 377. (doi :10.1016/j.biopsych.2013.05.040).
148
Widiger TA& Presnall JR. 2012 Application clinique du modèle à cinq facteurs. J. Pers. 81, 515 à 527. (doi :10.1111/jopy.12004). ,
149
Samuel DB et Gore WL. 2012 Variantes inadaptées de la conscience et de l’agréabilité. J. Pers. 80, 1669-1696. (doi :10.1111/j.1467-6494.2012.00770.x).
150
Widiger TA& Costa PT. 2012 Intégration de la structure normale et anormale de la personnalité : le modèle à cinq facteurs. J. Pers. 80, 1471 à 1506. (doi :10.1111/j.1467-6494.2012.00776.x).
151
Bartz J, Simeon D, Hamilton H, Kim S, Crystal S, Braun A, Vicens V & Hollander E. 2011 L’ocytocine peut entraver la confiance et la coopération dans le trouble de la personnalité limite. Soc. Cogn. Affect. Névrose. 6, 556 à 563. (doi :10.1093/scan/nsq085).
152
Dinsdale N& Crespi BJ. 2013 Le paradoxe de l’empathie limite : preuves et modèles conceptuels pour les améliorations empathiques dans le trouble de la personnalité limite. J. Pers. Disord. 27, 172 à 195. (doi :10.1521/pedi.2013.27.2.172).
153
Zahn-Waxler C, Crick N, Shirtcliff EA et Woods K. 2006 Les origines et le développement de la psychopathologie chez les femmes et les hommes. Psychopathologie du développement, vol. 1 (eds, Cicchetti D& Cohen DJ), pp. 76-138. Hoboken, NJ : Wiley.
154
Zahn-Waxler C, Shirtcliff EA& Marceau K. 2008 Troubles de l’enfance et de l’adolescence : genre et psychopathologie. Annu. Rev. Clin. Psychol. 4, 275 à 303. (doi :10.1146/annurev.clinpsy.3.022806.091358).
155
Samuel DB, Carroll KM, Rounsaville BJ& Ball SA. 2013 Troubles de la personnalité en tant que variantes inadaptées et extrêmes de la personnalité normale : trouble de la personnalité limite et névrosisme dans une substance utilisant un échantillon. J. Pers. Disord. 27, 625 à 635. (doi :10.1521/pedi.2013.27.5.625).
156
Haig, D., 2003 Sur la réciprocité intrapersonnelle. Evol. Hum. Behav. 24, 418 à 425. (doi :10.1016/S1090-5138(03)00063-1). ,
157
Crespi, B& Badcock, C., 2008 Psychose et autisme en tant que troubles diamétralement du cerveau social. Se comporter. Sci. cérébrale 31, 241 à 261. (doi :10.1017/S0140525X08004214).
158
Tsang SY, et al. 2013 Rôle cognitif social du gène candidat de la schizophrénie GABRB2. PLoS ONE 8, e62322. (doi :10.1371/journal.pone.0062322).
159
Ubeda F& Gardner A. 2010 Un modèle d’empreinte génomique dans le cerveau social : les juvéniles. Évolution 64, 2587 à 2600. (doi :10.1111/j.1558-5646.2010.01015.x).
160
Francks C, et al. 2007 LRRTM1 sur le chromosome 2p12 est un gène maternel supprimé qui est associé paternellement à la gaucherie et à la schizophrénie. Mol. Psychiatry 12, 1129-1139. (doi :10.1038/sj.mp.4002053).
161
Queller DC. 2011 Élargissement de la condition physique sociale et règle de Hamilton pour la parenté, le kith et la gentillesse. Proc. Natl Acad. Sci. États-Unis 108, 10 792–10 799. (doi :10.1073/pnas.1100298108). ,
162
Frank SA. 2003 Répression de la concurrence et évolution de la coopération. Évolution 57, 693 à 705. PubMed,
163
Boehm, C& Flack, J., 2010 L’émergence de structures de pouvoir simples et complexes à travers la construction de niches sociales. La psychologie sociale du pouvoir (éd. & Guinote A), pp. 46-87. Londres, Royaume-Uni : Guilford.
164
Nesse RM. 2004 La sélection naturelle et l’insaisissabilité du bonheur. Phil. Trans. R. Soc. Lond. B 359, 1333 à 1347. (doi :10.1098/rstb.2004.1511).
165
Keller MC& Nesse RM. 2005 La mauvaise humeur est-elle une adaptation ? Preuves de sous-types dont les symptômes correspondent à ceux des facteurs déclenchants. J. Affecter. Disord. 86, 27 à 35. (doi :10.1016/j.jad.2004.12.005).
166
Ortie D. 2004 Origines évolutives de la dépression : une revue et une reformulation. J. Affecter. Disord. 81, 91 à 102. (doi :10.1016/j.jad.2003.08.009).
167
Andrews PW et Thomson JA. 2009 Le bon côté d’être bleu : la dépression comme adaptation à l’analyse de problèmes complexes. Psychol. Rev. 116, 620 à 654. (doi :10.1037/a0016242).
168
Johnson SL, Fulford D& Carver CS. 2012 L’épée à double tranchant de l’engagement d’objectif : conséquences de la poursuite d’un objectif dans le trouble bipolaire. Clin. Psychol. Psychother. 19, 352 à 362. (doi :10.1002/cpp.1801).
169
Berridge KC, Robinson TE et Aldridge JW. 2009 Disséquer les composantes de la récompense : « aimer », « vouloir » et apprendre. Curr. Opin. Pharmacol. 9, 65 à 73. (doi :10.1016/j.coph.2008.12.014).
170
Segerstrale U. 2013 L’oracle de la nature : la vie et l’œuvre de W. D. Hamilton. Oxford, Royaume-Uni : Oxford University Press.
Et pour aller plus loin
Profil de nos 5000 abonnés
| Par catégorie professionnelle | |
| Médecins | 27% |
| Professions de santé | 33% |
| Sciences de la vie et de la terre | 8% |
| Sciences humaines et sociales | 12% |
| Autres sciences et techniques | 4% |
| Administration, services et tertiaires | 11% |
| Economie, commerce, industrie | 1% |
| Médias et communication | 3% |
| Art et artisanat | 1% |
| Par tranches d'âge | |
| Plus de 70 ans | 14% |
| de 50 à 70 ans | 53% |
| de 30 à 50 ans | 29% |
| moins de 30 ans | 4% |
| Par motivation | |
| Patients | 5% |
| Proche ou association de patients | 3% |
| Thèse ou études en cours | 4% |
| Intérêt professionnel | 65% |
| Simple curiosité | 23% |
Si vous n'êtes pas abonné
C'est ici
INUTILE si vous vous êtes déjà abonné, car vous le restez tant que vous ne demandez pas votre désisncription
Médecine évolutionniste (ou darwinienne)
Depuis quelques années, le problème de l'antibiorésistance, les progrès de la génomique, la redécouverte du microbiote et la prise en charge de maladies au long cours, nécessitent l'introduction d'une pensée évolutionniste dans la réflexion clinique.
Le premier diplôme universitaire intitulé "Biologie de l'évolution et médecine" a été mis en place à la faculté de Lyon en 2016.
Vous aimerez aussi ces humeurs...
Les spécialistes de la prévalence - Une hypothèse concerne l’avenir, un fait concerne le passé. Entre les deux il y a [...]
Déclin de la transcendance - La neurophysiologie fascine par ses progrès fulgurants et agace par sa prétention à percer [...]
Rendons des malades à Pluton - Trier, classer, ordonner, catégoriser sont certainement les activités primaires de tout [...]
Valse des normes - Dans les années 1970, la pression artérielle systolique à 60 ans ne devait pas dépasser 160 [...]
LUCA et le Big Bang - Depuis Galilée, qui avait tant agacé nos papes, la vulgarisation de l’astronomie a [...]
Médecine darwinienne ou évolutionniste
• Evolution : influence sur la santé et les maladies • Evolution : et génétique en [...]
Vous aimerez aussi...
Cancers et multicellularité - Le cancer existe depuis le début de la vie multicellulaire. Il y a environ un milliard [...]
LUCA : notre ancêtre universel - LUCA : la cellule qui a engendré toute la vie dur la terre L’image actuelle de notre « [...]
Génétique évolutionniste des maladies coronaires. - La susceptibilité aux maladies communes telles que les maladies coronaires reflète en partie [...]
ADN poubelle de l'intelligence humaine - Les traits qui séparent les chimpanzés des humains semblent évidents, nous sommes pourtant des [...]
Allaitement et évolution chez Homo sapiens - Allaitement maternel, laisser tomber les standards ? Les bénéfices apportés par l’allaitement [...]
Vous aimerez aussi...
Biais sexuel dans les maladies auto-immunes pédiatriques - Les maladies auto-immunes affectent jusqu'à 10% de la population mondiale et environ 80% des [...]
Reproduction sexuée face au cancer - Abstract L'origine et le maintien ultérieur du sexe et de la recombinaison sont parmi les [...]
Coévolution gènes-culture et ligne Hajnal - La ligne allant de Trieste à St Pétersbourg est nommée ligne Hajnal suite aux travaux de John [...]
Culture cumulative et gros cerveau - Au cours des derniers millions d'années, la taille du cerveau des hominidés a plus que [...]
Évolution de notre espèce depuis 2000 ans - Une question est fréquemment posée aux biologistes de l’évolution : "Les humains évoluent-ils [...]
La phrase biomédicale aléatoire
Le type d'environnement nécessaire à telle époque particulière de la vie d'un être détermine, en un sens, le type d'objet qui va émerger à un moment donné sur son terrain de vie. Et s'il n'émerge pas au moment adéquat, il est considéré comme étant insignifiant ou non existant.
― Kinji Imanishi (philosophe de l'évolution)