
Influence de l’histoire évolutionniste sur la santé et les maladies
dernière mise à jour le 12/02/2021
En glissant avec la souris sur les mots surlignés, leur explication apparaît.
Abstract
Presque tous les variants génétiques qui influencent le risque de maladie ont des origines spécifiques à l'homme ; cependant, les systèmes qu'ils contrôlent ont des racines anciennes qui remontent souvent à des événements bien antérieurs à l'origine des humains. Ici, nous passons en revue comment les progrès dans notre compréhension des architectures génétiques des maladies, l'évolution humaine récente et l'histoire évolutionniste profonde peuvent aider à expliquer comment et pourquoi les humains deviennent malades dans les environnements modernes. Les populations humaines présentent des différences dans la prévalence de nombreuses maladies génétiques courantes et rares. Ces différences sont en grande partie le résultat de la diversité des histoires environnementales, culturelles, démographiques et génétiques des populations humaines. Synthétiser nos connaissances croissantes de l’histoire de l’évolution avec la médecine génétique, tout en tenant compte des facteurs environnementaux et sociaux, aidera à une génomique personnalisée et à découvrir le potentiel caché dans l’ADN de chaque individu pour guider les décisions cliniques. En bref, la médecine de précision est fondamentalement une médecine évolutionniste, et l'intégration de perspectives évolutionnistes dans la clinique en révèle le potentiel.
Introduction
Une maladie génétique est un produit inévitable et nécessaire de l'évolution (encadré 1). Les systèmes biologiques fondamentaux, tels que la réplication, la transcription et la traduction de l'ADN, ont évolué très tôt dans l'histoire de la vie. Ces anciens processus ont donné naissance à la vie cellulaire, mais aussi créé un potentiel de maladie. Les innovations ultérieures tout au long de l’histoire évolutionniste ont également permis à la fois l’adaptation et le potentiel de dysfonctionnement. Dans ce contexte ancien, de jeunes variants génétiques spécifiques à la lignée humaine interagissent avec les environnements modernes pour produire des phénotypes de maladies humaines. Par conséquent, les substrats des maladies génétiques chez les humains modernes sont souvent bien plus anciens que la lignée humaine elle-même, mais les variants génétiques qui les provoquent sont généralement spécifiques aux humains.
L'avènement des technologies génomiques à haut débit a permis le séquençage des génomes de diverses espèces dans l'arbre de vie [1]. L'analyse de ces génomes a, à son tour, révélé la conservation frappante de nombreuses voies moléculaires qui sous-tendent la fonction des systèmes biologiques essentiels à la vie cellulaire [2]. Les mêmes technologies ont aussi révolutionné la génomique humaine [3] ; actuellement, plus de 120 000 séquences individuelles de génomes humains entiers sont accessibles au public, et des données à l'échelle du génome provenant de centaines de milliers d'autres ont été générées par des sociétés de génomique grand public [4]. D'énormes biobanques nationales caractérisent également les génotypes et phénotypes de millions de personnes du monde entier [5, 6, 7]. Ces études changent radicalement notre compréhension de l' architecture génétique de la maladie [8]. Il est aussi désormais possible d'extraire et de séquencer de l'ADN ancien à partir de restes d'organismes vieux de plusieurs milliers d'années, permettant de reconstituer l'histoire de l'adaptation humaine récente avec une résolution sans précédent [9, 10]. Ces avancées ont révélé l'histoire récente, souvent compliquée, de notre espèce et son influence sur l'architecture génétique des maladies [8, 11]. Avec l'expansion du séquençage clinique du génome entier et de la médecine personnalisée, l'influence de notre passé évolutionniste et ses implications pour la compréhension des maladies humaines ne peuvent plus rester ignorées par la pratique médicale; les perspectives évolutionnistes doivent informer la médecine [12, 13].
Tout comme l’histoire médicale d’une famille au fil des générations, le génome est un document historique. Le décodage de l'évolution du génome humain fournit est précieux pour interpréter et modéliser les maladies. Ceci ne se limite pas à l’évolution humaine récente, mais comprend également des événements plus anciens qui couvrent l’histoire de la vie. Dans cette revue, nous retraçons 4 milliards d’années d’interactions entre l’évolution et les maladies, en illustrant comment les innovations de l’histoire de la vie ont établi le potentiel et l’inévitabilité des maladies. En commençant par des événements très lointains d’où proviennent la plupart des voies et des gènes impliqués dans la maladie humaine, nous expliquons comment les systèmes biologiques anciens, les variants génétiques récents et les environnements dynamiques interagissent pour produire à la fois l’adaptation et les risques de maladies. Nous ne pouvons évidemment pas fournir un compte rendu complet de tous les événements évolutionnistes pertinents pour nos maladies. Notre objectif est plutôt d'illustrer par des exemples la pertinence de l'évolution profonde et récente pour l'étude et le traitement des maladies génétiques. Bon nombre de ces informations clés proviennent de découvertes récentes, qui n'ont pas encore été intégrées dans le canevas plus large de la biomédecine évolutionniste (encadré 2).
Encadré 1 : Inévitables tenants évolutionnistes de la maladie
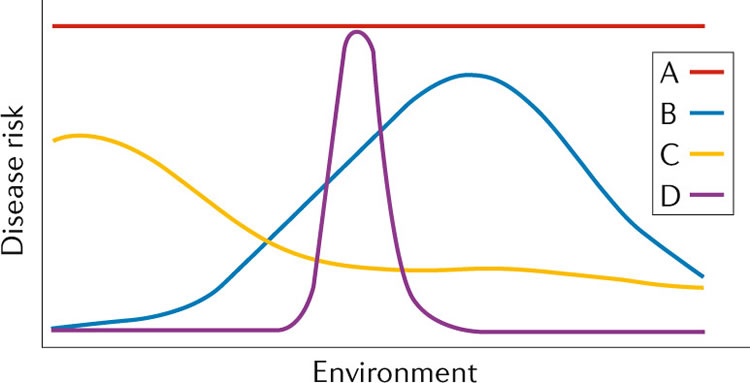
La définition de la maladie varie selon les perspectives biologiques, médicales et évolutionnistes. Voir la maladie à travers le prisme de l'évolution fournit un cadre flexible et puissant pour définir et classer les maladies [12]. Comme illustré dans les normes de réaction tracées sur la figure ci-dessous, le risque de maladie (axe y) est une fonction à la fois du génotype (lignes colorées) et de l'environnement (axe x). Certains génotypes mènent à des maladies dans tous les environnements (lignée A) ; les troubles mendéliens à haute pénétrance appartiennent à ce groupe. À l'autre extrême, le risque de maladie ne peut survenir que dans le cas d'un couple très spécifique d'environnement et de génotype (lignée D). La phénylcétonurie (PKU), qui se manifeste uniquement en présence de mutations qui rendent à la fois les copies de l'enzyme phénylalanine hydroxylase non fonctionnelles et un régime comprenant de la phénylalanine, illustre ce cas. La plupart des maladies se situent entre ces extrêmes (lignes B et C). La maladie résulte souvent de discordances entre le génotype et l’environnement. Par exemple, le risque élevé d'obésité, une maladie chronique à héritabilité importante (30 à 40%) [177], dans de nombreuses populations modernes est dû (au moins en partie) à des changements rapides et récents de nos modes de vie [178], comme alimentation plus calorique, sédentarité et sommeil plus court. Ici, l’obésité se manifeste en raison d’une inadéquation entre le génotype et un environnement en évolution rapide.
Les génotypes ont souvent des effets opposés sur différents traits. Ce phénomène de pléiotropie antagoniste, conduit souvent à des maladies [179]. L’exemple type est la sélection équilibrée entre le variant HBB de l’hémoglobine qui protège contre le paludisme mais conduit à l'anémie falciforme. La pléiotropie antagoniste a également été détectée dans des traits génétiques complexes, tels que les maladies cardiaques, où les allèles qui augmentent le succès reproducteur à vie augmentent également le risque de maladie cardiaque [180]. Ainsi, de nombreuses maladies actuelles existent parce que les populations ne se sont pas adaptées à des environnements changeants ou que des adaptations antérieures conduisent à des compromis entre santé et valeur sélective. Cependant, la maladie n'est pas seulement un produit du monde moderne. Tant qu'il y a une variation phénotypique, la maladie est inévitable; certains individus seront mieux adaptés à certains environnements, et donc plus sains que d'autres.
Encadré 2 : Médecine évolutionniste
La médecine évolutionniste est l'étude de l’impact de l’évolution sur les traits humains et les maladies humaines et comment la biologie de l’évolution peuvent être introduite en médecine. Cette revue se concentre sur les apports récents de la génomique pour cette médecine. Mais elle est bien plus vaste et a été développée ailleurs [12,26,181]. En voici néanmoins les grands principes qui reposent sur l'idée que les maladies humaines émergent des contraintes, des compromis, des inadéquations et des conflits inhérents à des systèmes biologiques complexes interagissant (via la sélection naturelle) avec des environnements divers et changeants (encadré 1).
La médecine évolutionniste a identifié plusieurs catégories d'explications pour les maladies génétiques complexes. La première est que la sélection naturelle n'aboutit pas à des corps parfaits mais opère avant tout sur l’aptitude reproductive relative contrainte par les lois de la physique et le rôle, la disponibilité et les interactions des variations biologiques préexistantes qui façonnent ou contraignent le cours ultérieur de l'évolution [182, 183]. La deuxième catégorie est l'inadéquation entre notre héritage biologique et nos environnements modernes [184]. L'inadéquation entre nos adaptations biologiques aux environnements ancestraux et les modes de vie modernes contribue à de nombreuses maladies courantes, telles que l'obésité, le diabète et les maladies cardiaques, qui sont favorisées par des modes de vie sédentaires et une mauvaise alimentation [185, 186]. Par exemple, une exposition passée à des conditions de famine ou de peu d’apport calorique peut favoriser des variants de gènes «économes» métaboliquement efficaces qui conduisent à l’obésité dans des environnements riches en calories. La troisième catégorie est celle des compromis, c’est l'idée qu'il existe des combinaisons de caractères qui ne peuvent pas être optimisées simultanément par la sélection naturelle [50, 51]. Le concept de compromis est lié à la contrainte évolutionniste, mais englobe un large ensemble de phénomènes qui façonnent l'évolution des traits. Par exemple, de nombreux traits liés à la valeur sélective s'appuient sur des réserves énergétiques communes, et l'investissement dans l'un se fait au détriment d'un autre [52]. De même, les variants génétiques pléiotropes qui influencent plusieurs systèmes conduisent à un compromis. De plus, les symptômes qui sont interprétés comme une maladie peuvent en fait représenter des réponses adaptatives conditionnelles. Enfin, les conflits évolutionnistes fournissent une quatrième catégorie d’explications possibles. Tous les organismes multicellulaires sont des agrégats de gènes et de génomes avec des histoires évolutionnistes différentes et des intérêts stratégiques divers. Cela signifie que tous les traits exprimés par les métazoaires complexes sont un compromis équilibré entre différents éléments génétiques des organes et systèmes corporels [187]. La pathologie peut alors résulter d'un conflit lorsque les conditions perturbent ces compromis.
Empreintes macroévolutionnistes sur les maladies humaines
Les systèmes impliqués dans la maladie ont des origines anciennes
De nombreux systèmes et processus biologiques essentiels de la vie cellulaire, tels que la réplication, la transcription et la traduction de l’ADN, représentent d’anciennes innovations partagées par tous les organismes vivants. Bien qu'essentielle, chacune d’elles a généré des facteurs de maladies actuelles (Fig.1). Dans cette section, nous fournissons des exemples de la façon dont plusieurs innovations anciennes ont créé des substrats de dysfonctionnements et de maladies, et comment la prise en compte de ces histoires contribue à la compréhension de modèles et systèmes biologiques de maladies.
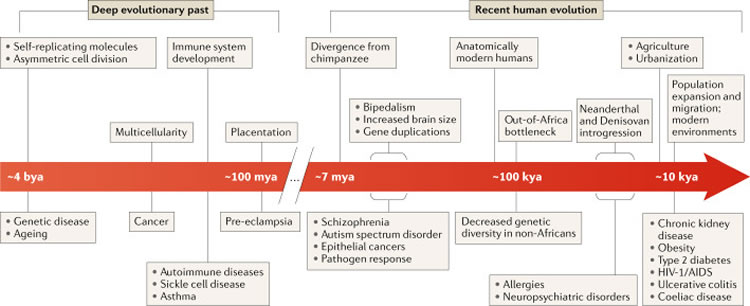
Figure 1 : Les événements évolutionnistes du passé profond et l'évolution humaine récente façonnent le potentiel de maladie.
Liste chronologique des événements du passé évolutionniste profond et celui de la lignée humaine (en haut) qui sont pertinents pour les modèles de risque de maladie humaine (en bas). Les anciennes innovations de cette chronologie (à gauche) ont formé des systèmes biologiques qui sont essentiels, mais qui sont également les fondements de la maladie. Au cours de l'évolution humaine récente (à droite), le développement de nouveaux caractères et les récents changements démographiques et environnementaux rapides ont créé le potentiel de conflits entre génotypes et environnements modernes qui peuvent causer des maladies.
La chronologie est schématique et non montrée à l'échelle. Bya : milliards d'années, Mya : millions d'années, Kya : milliers d’années.
À titre d'exemple fondamental (et évident), l'origine des molécules auto-réplicantes il y a 4 milliards d'années a formé la base du vivant, mais aussi la racine des maladies génétiques [12,14,15]. De même, la division cellulaire asymétrique peut avoir évolué comme un moyen efficace de gérer les dommages cellulaires, mais elle a également établi les bases du vieillissement dans les organismes multicellulaires [16,17]. Une myriade de maladies liées à l'âge chez l'homme et de nombreux autres organismes multicellulaires sont une manifestation de ce premier compromis évolutif.
L'évolution de la multicellularité, qui s'est produite à plusieurs reprises dans l'arbre de la vie, illustre les interactions entre innovation évolutionniste et maladie [18]. L'origine de la multicellularité a permis des plans corporels complexes avec des milliards de cellules, impliquant des innovations associées à la capacité des cellules à réguler leurs cycles, à moduler leur croissance et à former des réseaux de communication complexes. Mais la multicellularité a également jeté les bases du cancer [19, 20]. Les gènes qui régulent le contrôle du cycle cellulaire sont souvent divisés en deux groupes: les caretakers (gardiens du génome) et les gatekeepers (gardes-barrières) [21, 22].
Les caretakers permettent à la cellule de transmettre à l’identique son patrimoine génétique lors des divisions cellulaires successives. Ils sont impliqués dans le contrôle de base du cycle cellulaire et la réparation de l'ADN. Les mutations de ces gènes entraînent souvent une augmentation des taux de mutations ou une instabilité génomique, qui augmentent le risque de cancer. Les caretakers sont enrichis pour des fonctions dont les origines remontent aux premières cellules [23].
Les gatekeepers régulent l’entrée de la cellule dans les différentes phases du cycle cellulaire. Ils sont apparus plus tard, lors de la genèse de la multicellularité des métazoaire [23]. Ils sont directement liés à la tumorigenèse à travers leurs rôles régulateurs dans la croissance, la mort et la communication cellulaires. La progression des tumeurs individuelles chez un patient donné répond aussi aux lois de l’évolution. Les nouveaux traitements du cancer tiennent compte de ces processus darwiniens que sont l'évolution de la résistance aux médicaments et l'hétérogénéité des clones cellulaires tumoraux [24, 25, 26, 27, 28, 29].
Comme la multicellularité, l'évolution des systèmes immunitaires a également ouvert la voie à la dérégulation et à la maladie. Les systèmes immunitaires innés et adaptatifs des mammifères sont tous deux anciens. Les composants du système immunitaire inné sont présents dans les métazoaires et même dans certaines plantes [30, 31], tandis que le système immunitaire adaptatif est présent chez les vertébrés à mâchoires [32]. Ces systèmes ont fourni des mécanismes moléculaires pour la reconnaissance du moi et du non-moi et la réponse aux agents pathogènes, mais ils ont évolué de manière fragmentaire, en utilisant de nombreux gènes et processus différents et préexistants. Par exemple, la cooptation de rétrovirus endogènes a fourni de nouveaux éléments de régulation pour la réponse à l'interféron [33]. De plus, il est clair que le système immunitaire humain a co-évolué avec des parasites, comme les helminthes, au cours de millions d'années. L'infection par les helminthes induit et module la réponse immunitaire chez l'homme [34].
Des analyses évolutionniste du développement ont révélé que de nouvelles structures anatomiques surgissent souvent en cooptant des structures et des voies moléculaires préexistantes. Par exemple, les yeux des animaux, la structure des membres chez les tétrapodes et la grossesse chez les mammifères (encadré 3) ont chacun évolué en adaptant et en intégrant des gènes anciens et en modifiant des circuits de régulation [35, 36, 37, 38]. Cette intégration de caractères nouveaux dans le réseau existant de systèmes biologiques établit de nouveaux ponts entre divers traits via les gènes partagés qui sous-tendent leur développement et leur fonction [36]. En conséquence, de nombreux gènes sont pléiotropes : ils ont des effets sur de multiples traits apparemment sans rapport. Nous ne pouvons pas développer ici toute la portée évolutionniste de ces innovations et de leurs héritages, mais comme dans chacun des cas décrits ci-dessus, des innovations et des adaptations allant de l'origine des métazoaires aux populations humaines modernes ont façonné autant de substrats pour de nouvelles maladies.
Encadré 3 : La grossesse comme étude de cas en médecine évolutionniste
La grossesse chez les mammifères illustre comment la prise en compte de l’histoire d’un trait à travers l’évolution peut éclairer notre compréhension de la maladie. Tous les humains qui ont vécu ont fait l’expérience de la grossesse, mais sa complexité est remarquable, elle implique la coordination entre plusieurs génomes et l'intégration physiologique entre des individus, elle est gérée par un organe transitoire, le placenta [188]. De plus, en assurant la transmission générationnelle de l'information génétique, elle fournit le substrat de toute l’évolution et le renouvellement de la vie elle-même [189].
Aspects macroévolutionnistes de la grossesse
La grossesse chez les mammifères placentaires, apparue il y a environ 170 millions d'années, implique l'intégration physiologique des tissus fœtaux et maternels via le placenta, un organe extra-embryonnaire transitoire dérivé du fœtus. La naissance vivante et la placentation ouvrent la porte à des interactions entre la mère et le fœtus pour l'approvisionnement en ressources, avec un conflit potentiel, car la mère a tendance à en fournir au fœtus moins que ce qu’il demande, elle a en effet d’autres besoins énergétiques, dont celui de prendre soin d’autres enfants nés ou à naître. Chez certains mammifères, y compris les humains, la placentation est très invasive, mettant en place une lutte physiologique entre la mère et le fœtus pour l'approvisionnement. Lorsque cet équilibre précaire est perturbé, des maladies de grossesse peuvent survenir. Un mauvais remodelage artériel maternel pendant la placentation limite l'efficacité placentaire, ce qui provoque une réponse compensatoire du fœtus en détresse. Ce déséquilibre entraîne une inflammation, une hypertension, des lésions rénales et une protéinurie chez la mère, ainsi qu'une augmentation du stress oxydatif et une naissance prématurée [190]. C’est la pré-éclampsie d’origine vasculaire, avec un mauvais pronostic pour la mère et le fœtus en l'absence de traitement. Comprendre la pré-éclampsie comme résultat d'un bras de fer évolutionniste entre la mère et le fœtus a des implications médicales [191, 192, 193, 194].
Aspect spécifique à Homo sapiens
Le moment de la naissance est la clé d'une grossesse saine et réussie, mais on en sait peu sur les mécanismes qui régissent et déclenchent le début de la parturition. La progestérone et ses récepteurs sont impliqués dans la parturition chez toutes les espèces vivipares ; cependant, la manière dont la progestérone régule la parturition peut être spécifique à l'espèce. Par exemple, le récepteur de la progestérone humaine (PGR) a présenté une évolution rapide après la divergence du dernier ancêtre commun avec les chimpanzés [195, 196]. Il existe des différences fonctionnelles entre les versions humaine et néandertalienne du récepteur de la progestérone [197]. Les changements spécifiquement humains du PGR influencent sa transcription et probablement sa phosphorylation [198, 199]. De même, les loci associés à la naissance prématurée humaine ont subi diverses forces évolutionnistes, dont une sélection équilibrée, une sélection positive et des différenciations entre populations [200]. Les types rapides et divers de changements évolutionnistes observés dans le PGR et certains des loci associés à la naissance prématurée rendent difficile leur extrapolation expérimentale à des modèles animaux, tels que les souris. De plus, les humains et les souris diffèrent dans les stratégies de reproduction, la morphologie de l'utérus, la placentation, la production d'hormones et les facteurs d'activation utérine [201]. Par exemple, la progestérone est produite par la mère chez la souris tout au long de la grossesse, tandis que chez l'homme, sa production se déplace vers le placenta après les premiers stades de grossesse. Compte tenu de l'histoire évolutionniste unique de la grossesse humaine, de nombreux aspects moléculaires de la grossesse peuvent être mieux étudiés dans d'autres organismes modèles, voire dans des systèmes à base de cellules humaines.
Au niveau des populations humaines
L’énigme centrale de la grossesse chez les mammifères est que le système immunitaire maternel ne rejette pas le fœtus étranger ; au contraire, il a non seulement évolué pour accepter le fœtus, mais il est également essentiel dans le processus de placentation [202, 203]. La centralité du système immunitaire maternel pendant la grossesse a des implications médicales importantes. Sa modulation se traduit par une capacité réduite à éliminer certaines infections [204, 205]. Les cellules tueuses naturelles utérines (uNK) et leurs récepteurs inhibiteurs de cellules tueuses (KIR) coopèrent avec les trophoblastes fœtaux pour réguler la réponse immunitaire maternelle. En outre, les cellules uNK sont également impliquées dans la réponse immunitaire aux agents pathogènes, et ce double rôle aboutit à des compromis évolutionnistes. Par exemple, l'haplotype KIR AA spécifiquement humain est associé à un poids de naissance inférieur et à une pré-éclampsie, ainsi qu'à une défense plus efficace contre le virus Ebola et de l'hépatite [206, 207] (Fig. 4a). Les populations humaines modernes ont des variations dans la diversité et l'identité des haplotypes KIR, probablement en raison de la sélection sur la placentation et la défense de l'hôte [208]. Les flambées de maladies infectieuses exercent donc une pression sélective uniquement en cours de grossesse. Mais les maladies infectieuses chroniques et sévères, telles que le paludisme, produisent souvent des changements significatifs dans la fréquence des allèles au niveau de la population dans les gènes liés à la grossesse, tels que FLT1 dans des populations de zones d'endémie palustre en Tanzanie [209]. La variabilité de pression des maladies infectieuses contribue à une variabilité du risque de maladies liées à la grossesse dans les populations modernes.
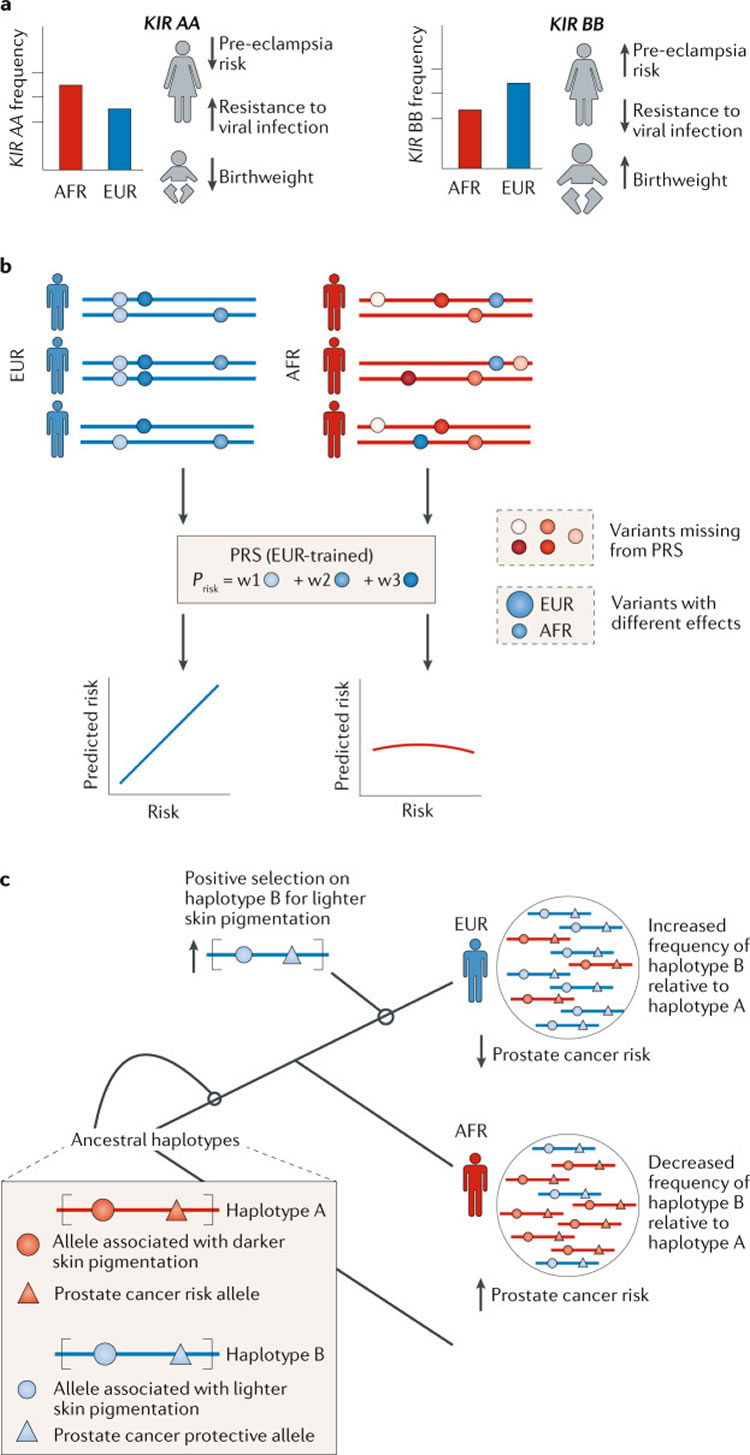
Figure 4 : Illustrations de la nécessité de prendre en compte diverses populations humaines dans l'analyse génétique des maladies.
a/ Les interactions entre le génotype du récepteur inhibiteur des cellules tueuses maternelles (KIR) et les trophoblastes fœtaux illustrent les compromis évolutifs pendant la grossesse. Le poids à la naissance est sous sélection stabilisatrice dans les populations humaines. L'interaction entre les génotypes maternels KIR (dont une diversité est maintenue dans la population) et les trophoblastes fœtaux, influencent le poids à la naissance. Les populations africaines (AFR), par rapport aux populations européennes (EUR), maintiennent des proportions plus importantes de l'haplotype KIR AA [176], ce qui est associé à une meilleure réponse immunitaire maternelle à certains défis viraux ; cependant, il est également associé à un faible poids à la naissance. Alternativement, l'haplotype KIR BB est associé à un poids de naissance plus élevé mais à un risque accru de pré-éclampsie.
b/ Les stratégies actuelles de prédiction du risque génétique sont perturbées par le manque d'inclusion de diverses populations humaines dans les études. Ainsi, les populations sous-représentées dans les bases de données génétiques, diminuent la prédiction des risques génétiques. Par exemple, les modèles de score de risque polygénique (PRS) établis sur les populations européennes donnent souvent de mauvais résultats lorsqu'ils sont appliqués aux populations africaines. Cette mauvaise performance provient du fait que la diversité génétique des populations africaines, les différences de taille d'effet entre les populations et les pressions évolutionnistes différentielles ne sont pas prises en compte. Les poids de chaque variante (cercles bleus) dans le PRS dérivés d'études d'association à l'échelle du génome sont signifiés par w1, w2 et w3.
c/ L'adaptation propre à la population et l'auto-stop génétique peuvent produire des risques différents selon les populations. Les haplotypes ayant des effets protecteurs contre une maladie peuvent atteindre une fréquence élevée dans des populations spécifiques par auto-stop génétique, entrainant des allèles proches, mais codant un caractère différent. Par exemple, la sélection pour une faible pigmentation a logiquement augmenté la fréquence d'un haplotype portant un variant associé à une peau plus claire (cercle bleu) dans les populations européennes par rapport aux populations africaines. Cet haplotype portait également une variante protectrice contre le cancer de la prostate (triangle bleu).
Implications médicales
Bien que les anciennes innovations macroévolutionnistes puissent sembler très éloignées des phénotypes humains modernes, leur empreinte persiste sur le corps et le génome humains. Comprendre les contraintes qu'ils imposent peut donner un aperçu de certains processus pathologiques.
Cartographier les origines et l'évolution des traits et identifier les réseaux génétiques qui les sous-tendent sont essentiels pour la sélection de modèles et l'extrapolation aux populations humaines. Le fait de ne pas prendre en compte l'histoire évolutionniste des systèmes homologues, leurs relations phylogénétiques et leurs contextes fonctionnels dans différents organismes peut conduire à une généralisation inexacte. Au lieu de cela, lorsqu'on considère un système modèle, des questions évolutionnistes clés sur l'organisme et sur le trait considéré peuvent indiquer dans quelle mesure la recherche sera traduisible pour les humains [39, 40]. Par exemple, la similitude entre un trait humain et un trait dans le système modèle est-elle due à une ascendance partagée, c'est-à-dire à une homologie ? La présence d'homologie dans un gène humain ou un système d'étude suggère la possibilité d’un système modèle ; cependant, l'homologie seule ne suffit pas. Les facteurs environnementaux et biologiques façonnent les traits, et la divergence entre les espèces complique l’hypothèse selon laquelle l'homologie conduit à une similitude génétique ou mécanique. Ainsi, l'homologie doit être complétée en essayant de comprendre si la divergence évolutionniste entre les humains et le modèle proposé conduit à une divergence fonctionnelle. Par exemple, l'évolution rapide du placenta et la variation de la stratégie de reproduction chez les mammifères ne permettent pas d’extrapoler sur la régulation du moment de la naissance, entre le modèle de la souris et les humains (encadré 3). Plus généralement, les différences dans les réseaux génétiques qui sous-tendent le développement de traits homologues chez les mammifères expliquent pourquoi la majorité des essais réussis sur les animaux ne se traduisent pas en essais cliniques humains [41, 42]. Les mécanismes moléculaires des systèmes anciens, tels que la réplication de l'ADN, peuvent être étudiés en utilisant des espèces phylogénétiquement éloignées; cependant, «humaniser» ces modèles pour rechercher des aspects de caractères spécifiques à l’homme peut être impossible sans d’autres études comparatives entre espèces étroitement apparentées [40].
Bien que la divergence évolutionniste des traits homologues soit un obstacle à la traduction directe des résultats d'un système modèle vers les humains, comprendre comment ces différences se sont produites peut également donner des informations sur les mécanismes d’une maladie. Par exemple, l'intuition suggérerait que les grands animaux à longue durée de vie (donc ayant de nombreuses cellules, divisions cellulaires et cellules vieillissantes), comme les éléphants et les baleines, courraient un risque accru de développer un cancer. Pourtant, la taille et la durée de vie ne sont pas significativement corrélées avec le risque de cancer, les éléphants et les baleines n'ont pas un risque plus élevé de développer un cancer [43, 44]. Pourquoi ? Des études récentes sur l'évolution des gènes impliqués dans la réponse aux dommages à l'ADN chez les éléphants ont révélé des mécanismes susceptibles de contribuer à la résistance au cancer. Un ancien pseudogène de facteur inhibiteur de la leucémie (LIF6) a retrouvé sa fonction chez l'ancêtre des éléphants modernes. Ce gène fonctionne en conjonction avec le gène suppresseur de tumeur TP53, dont le nombre de copies a augmenté chez les éléphants, réduisant leur risque de cancer malgré leur grande taille [45, 46]. Cela illustre un compromis de base du cycle biologique : la sélection a créé des mécanismes de suppression du cancer et de maintien somatique chez les grands vertébrés qui ne sont pas nécessaires chez les petits vertébrés à courte durée de vie. L'étude de ces paradoxes peut suggérer des cibles pour des interventions fonctionnelles.
Évolution spécifique à l'homme
Adaptation humaine, compromis et maladie
Les événements macroévolutionnistes décrits ci-dessus ont créé le fondement des maladies génétiques, mais compte tenu des changements plus récents survenus au cours de l'histoire évolutionniste de la lignée humaine, il est nécessaire d'éclairer le contexte complet des maladies humaines. Des comparaisons entre les humains et leurs plus proches parents primates vivants, tels que les chimpanzés, ont révélé des maladies qui, soit n'apparaissent pas chez d'autres espèces, soit suivent des parcours très différents [47]. Nous commençons à comprendre les différences génétiques sous-jacentes à certaines de ces affections spécifiques à l'homme, avec un aperçu particulier pour les maladies infectieuses.
Les derniers ancêtres communs des humains et des chimpanzés ont subi un événement de spéciation complexe qui a probablement impliqué plusieurs cycles de flux de gènes entre 12 et 6 millions d'années (mya) [48]. Au cours des millions d'années qui ont suivi cette divergence, les pressions climatiques, démographiques et sociales ont entraîné l'évolution de nombreux traits physiques et comportementaux propres à la lignée humaine, notamment la bipédie (~ 7 mya), la disparition des poils corporels (~ 2–3 mya) et un plus grand volume cérébral par rapport à la taille (~ 2 mya) [12, 47]. Ces traits ont évolué dans un éventail diversifié de groupes d'hominidés, principalement en Afrique, bien que certaines de ces espèces, comme Homo erectus, se soient aventurées en Europe et en Asie.
Ces adaptations humaines se sont développées sur le substrat de systèmes étroitement intégrés façonnés par des milliards d'années d'évolution, et donc des adaptations bénéfiques pour un système ont souvent engendré des compromis sous forme de coûts pour d'autres systèmes liés [49]. Le concept de compromis dérive d'une branche de la biologie évolutionniste connue sous le nom de théorie des traits d'histoire de vie. Il est basé sur l'observation que les organismes contiennent des combinaisons de caractères qui ne peuvent pas être optimisées simultanément par la sélection naturelle [50,51]. Par exemple, de nombreux traits liés à la valeur sélective (fitness) s'appuient sur des réserves énergétiques communes, et l'investissement dans l'un se fait au détriment d'un autre [52]. Une grande taille corporelle peut améliorer la survie dans certains environnements, mais cela peut être compensé par un développement plus lent ou un moindre investissement dans la reproduction.
Le concept de compromis est cliniquement pertinent car il évite la notion de phénotype ou de valeur sélective «optima» uniques pour un individu [49,53,54]. Compte tenu des interconnections profondes liées à l’évolution du corps humain, de nombreuses maladies sont étroitement liées, en ce sens que la diminution du risque pour l'une augmente le risque pour l'autre. Ces maladies opposées et les compromis qui les produisent sont les plus difficiles lorsqu'il y a concurrence au sein du corps pour des ressources limitées; par exemple, l'énergie utilisée pour la reproduction ne peut pas être utilisée pour la croissance, pour la fonction immunitaire ou d'autres processus de survie coûteux en énergie [54]. La base moléculaire de ces maladies diamétrales résulte souvent d'une pléiotropie antagoniste au niveau génétique : un variant a des effets contrastés sur plusieurs systèmes corporels. Dans les cas extrêmes, certaines maladies qui se manifestent bien après l'âge de procréation, par exemple la maladie d'Alzheimer, ne sont pas vues par la sélection, donc plus propices aux compromis avec des traits plus coûteux. Le cancer et les troubles neurodégénératifs présentent également ce schéma diamétral, où le risque de cancer est inversement associé à la maladie d'Alzheimer, la maladie de Parkinson et la maladie de Huntington. Cette association est supposée être médiée par des différences dans l'utilisation de l'énergie neuronale et des compromis dans les voies de la prolifération cellulaire et de l'apoptose [49]. De même, l'arthrose (dégradation du cartilage dans les articulations souvent accompagnée d'une densité minérale osseuse élevée) et l'ostéoporose (faible densité minérale osseuse) coexistent rarement. Cet aspect diamétral reflète, au moins en partie, des variations individuelles pour la transformation des cellules souches mésenchymateuses de la moelle osseuse en ostéoblastes ou en adipocytes [49, 55]. Dans un autre exemple, la sélection d’une réponse immunitaire robuste peut conduire à un risque accru de maladies auto-immunes et inflammatoires, en particulier lorsqu'elle est associée à de nouvelles inadéquations environnementales [49, 54]. D'autres exemples de compromis dans le corps humain se manifestant par un risque de diverses maladies, y compris les troubles psychiatriques ou rhumatismaux [49, 56].
Tout comme les adaptations évolutionnistes ont créé de nouveaux substrats de maladie, les pressions exercées sur la lignée humaine ont jeté les bases de capacités cognitives complexes, mais elles ont également potentialisé le risque de maladies neuropsychiatriques ou neurodéveloppementales. Par exemple, des variants structuraux génomiques ont permis des innovations fonctionnelles dans le cerveau grâce à l'émergence de nouveaux gènes [57 ,58, 59, 60]. De nombreuses duplications segmentaires spécifiques à l'homme influencent les gènes essentiels au développement du cerveau humain, tels que SRGAP2C et ARHGAP11B. Ces deux gènes fonctionnent dans le développement du cortex cérébral et ont pu participer à l'expansion du volume cérébral [61, 62, 63]. On suppose également que le NOTCH2NL spécifique à l'homme a évolué à partir d'un événement de duplication partielle et qui a majoré la corticogenèse [59,60]. Bien que ces variants structuraux soient probablement adaptatifs [58], ils peuvent également avoir prédisposé les humains aux maladies neuropsychiatriques et aux troubles du développement. La variation du nombre de copies dans la région proche du ARHGAP11B, en particulier une microdélétion à 15q13.3, est associée à un risque de déficience intellectuelle, de trouble du spectre autistique (TSA), de schizophrénie et d'épilepsie [58, 64]. Les duplications et délétions de NOTCH2NL et des régions environnantes sont impliquées respectivement dans la macrocéphalie et le TSA ou la microcéphalie et la schizophrénie [59]. Ces compromis jouent également au niveau du domaine des protéines. Par exemple, le domaine Olduvai (anciennement connu sous le nom DUF1220) est une séquence de 1,4 kb qui apparaît en 300 copies environ dans le génome humain. Chez l’homme, ce domaine a connu une forte augmentation du nombre de copies. Ces domaines font apparaître des tableaux de gènes en tandem dans la famille des gènes de breakpoint des neuroblastomes (NBPF) et ont été associés à la fois à une augmentation de la taille du cerveau et à des maladies neuropsychiatriques, y compris l'autisme et la schizophrénie [65]. Ces exemples suggèrent que l'organisation génomique de ces duplications spécifiques à l'homme peut avoir permis des changements également spécifiques dans le développement du cerveau tout en augmentant également la probabilité de réarrangements préjudiciables pour certaines maladies humaines [59, 64]. En outre, les régions génomiques associées aux maladies neuropsychiatriques ont connu une évolution accélérée spécifique à l'homme et une sélection positive récente, fournissant des preuves supplémentaires du rôle des pressions évolutionnistes récentes sur le risque de maladie [66, 67]. Les loci associés à la schizophrénie, par exemple, sont enrichis à proximité des HAR (human accelerated regions), ce qui ne s’observe pas chez les primates non humains [68]. La variation des HAR a également été associée au risque de TSA, peut-être en raison de perturbations de l'architecture des gènes de régulation [69].
Les systèmes immunitaires humains se sont adaptés en réponse aux changements de l'environnement et des modes de vie au cours des derniers millions d'années; cependant, l'évolution rapide du système immunitaire peut avoir rendu les humains vulnérables à certaines maladies, comme l'infection par le VIH-1. Un virus similaire, le virus de l'immunodéficience simienne (SIV), est trouvé chez les chimpanzés et d'autres primates, et des études au début des années 2000 ont mis en évidence des symptômes de type SIDA (principalement une réduction des cellules T CD4 +) chez les chimpanzés infectés par le SIV. Bien que les effets du SIV chez les chimpanzés reflètent certains des effets du VIH chez les humains [70], les chimpanzés captifs infectés par le VIH-1 ne développent généralement pas le SIDA et ont de meilleurs résultats cliniques. Ces différences sont influencées par l'évolution immunitaire spécifique à l'homme. Par exemple, les humains, et non les singes, ont perdu l'expression de plusieurs Siglecs (protéines de surface cellulaire liées aux acides sialiques) dans les lymphocytes T 71. À l'appui de cette hypothèse, les cellules T humaines à forte expression de Siglec-5 survivent plus longtemps après une infection par le VIH-1 [72]. En outre, il existe un rôle possible pour les Siglecs en évolution rapide dans d'autres maladies, telles que les cancers épithéliaux, qui affectent différemment les humains par rapport aux primates étroitement apparentés [73, 74]. Un autre changement immunitaire spécifique à l'homme est la suppression d'un exon de la CMAH (CMP-N-acétylneuraminic acid hydroxylase) conduisant à une différence dans les sialoglycanes de surface cellulaire humaine par rapport à d'autres grands singes [75, 76, 77]. Le changement de l'acide sialique humain en une terminaison d'acide N-acétylneuraminique (Neu5Ac), plutôt qu’en acide N-glycolylneuraminique (Neu5Gc), peut avoir été induit par la pression pour échapper à l'infection par Plasmodium reichenowi, un parasite qui lie Neu5Gc et cause le paludisme chez les chimpanzés. À l'inverse, la prévalence de Neu5Ac a probablement rendu les humains plus sensibles à l'infection par le parasite du paludisme Plasmodium falciparum, qui se lie à Neu5Ac78,79, ainsi qu’à une autre pathologie spécifique à l'homme : la fièvre typhoïde [80]. La toxine typhoïde se lie spécifiquement et est cytotoxique pour les cellules exprimant les glycanes Neu5Ac. Ainsi, la suppression du CMAH a probablement été sélectionnée sous la pression d’agents pathogènes, mais elle a permis à son tour d'autres maladies spécifiques à l'homme comme le paludisme et la fièvre typhoïde [81]. L'évolution rapide du système immunitaire humain crée un potentiel de variations et de maladies spécifiques [82, 83].
Implications médicales
Ces exemples de l'évolution humaine récente mettent en évidence l'interaction continue entre la variation génétique, l'adaptation et la maladie. Comprendre l'histoire évolutionniste des traits ainsi que l'étiologie des maladies apparentées peut aider à identifier et à évaluer les risques des traitements en raison des compromis. Par exemple, les stéroïdes ovariens ont des effets pléiotropes stimulant à la fois la croissance osseuse et la mitose dans les tissus mammaires pour mobiliser les réserves de calcium pendant la lactation [54]. Cependant, plus tard dans la vie, ce lien donne lieu à un compromis clinique. L'hormonothérapie substitutive chez les femmes ménopausées réduit le risque d'ostéoporose et de cancer de l'ovaire, mais aussi, en raison de ses effets sur les tissus mammaires, augmente le risque de cancer du sein. Étant donné le caractère commun du compromis entre le maintien et la prolifération, il ne s'agit là que de l'un des nombreux exemples de risque de cancer émergeant à la suite de compromis dans les systèmes immunitaire, reproducteur et métabolique [56, 84]. La grossesse est également riche en compromis cliniquement pertinents étant donné l'interaction entre plusieurs individus et génomes (mère, père et fœtus) avec des objectifs différents (encadré 3). Les compromis au niveau cellulaire ont également des implications médicales. Par exemple, la sénescence cellulaire est nécessaire et bénéfique à de nombreuses réponses corporelles essentielles, mais l'accumulation de cellules sénescentes est à la base de nombreux troubles liés au vieillissement. Ainsi, les individus ayant résolu différemment ces compromis peuvent avoir des âges «moléculaires» et «chronologiques» très différents [85].
Identifier ces compromis en étudiant la maladie et la réponse au traitement est d'un grand intérêt, mais cela est difficile pour plusieurs raisons : le nombre de combinaisons possibles de caractères à considérer est important ; de nombreux humains doivent avoir subi les effets négatifs ; et les données doivent être disponibles sur les deux caractères chez les mêmes individus. Ici, l'évolution associée aux biobanques offre une solution [5, 86, 87]. En considérant le contexte évolutif et les liens potentiels entre les traits, l'espace de recherche des compromis possibles peut être limité. Ensuite, les traits diamétraux peuvent être testés parmi les individus dans les données massives de santé des biobanques en réalisant des études d'association à l'échelle du phénome (PheWAS équivalent des GWAS pour les génomes) soit sur les traits, soit sur les loci en recherchant des relations inverses [88]. Les mécanismes sous-jacents aux associations observées pourraient alors être évalués dans des systèmes modèles et, s'ils sont validés, anticipés pour de futurs traitements humains.
En plus des compromis, les analyses évolutionnistes peuvent nous aider à identifier des cibles thérapeutiques pour des maladies uniquement humaines. Un petit sous-ensemble d'humains infectés par le VIH ne progresse jamais vers le SIDA, ce phénotype de résistance est généralement attribué à la génomique de l'hôte [89, 90, 91]. Identifier et comprendre les gènes qui contribuent à la non-progression est d'un grand intérêt dans le développement de vaccins et de traitements contre l'infection à VIH. Des études d'association pangénomique (GWAS) et des études fonctionnelles ont confirmé le rôle de la région de classe I du CMH, en particulier les molécules HLA-B*27 / B*57, dans la non-progression du VIH [92, 93, 94]. La génomique comparative avec les chimpanzés a identifié une molécule de chimpanzé CMH de classe I, fonctionnellement analogue à celle des non-progresseurs, qui contient des substitutions d'acides aminés qui changent l'affinité de liaison pour les zones conservées des virus VIH-1 et SIV. L'analyse évolutionniste de cette région suggère que ces substitutions sont le résultat d'un ancien balayage sélectif dans les génomes de chimpanzés qui ne s'est pas produit chez l'homme [95]. Cette analyse nous aide non seulement à comprendre comment les humains sont particulièrement sensibles à la progression du VIH, mais met également en évidence les variations fonctionnelles dans le CMH qui sont des cibles potentielles d'intervention médicale.
Histoire récente de la démographie humaine
La plupart des variants génétiques sont jeunes, mais ont des histoires diverses
L'histoire démographique complexe des humains modernes au cours des 200 000 dernières années a créé des différences dans l'architecture génétique et un risque de maladies spécifiques dans les populations. Grâce aux séquences génomiques de milliers d'humains de provenances diverses, nous pouvons comparer les informations génétiques au fil du temps et de la géographie pour mieux comprendre les origines et l'évolution des variants génétiques individuels et des populations humaines [96, 97, 98]. La grande majorité des variants génétiques humains ne sont pas partagés avec d'autres espèces [99]. Les événements démographiques tels que les goulots d'étranglement, l'introgression et l'expansion de la population ont façonné la composition génétique des populations humaines, tandis que l'évolution très rapide des nouveaux environnements humains et les adaptations consécutives ont créé un potentiel d'inadéquations évolutionnistes (Fig. 2 et 3).
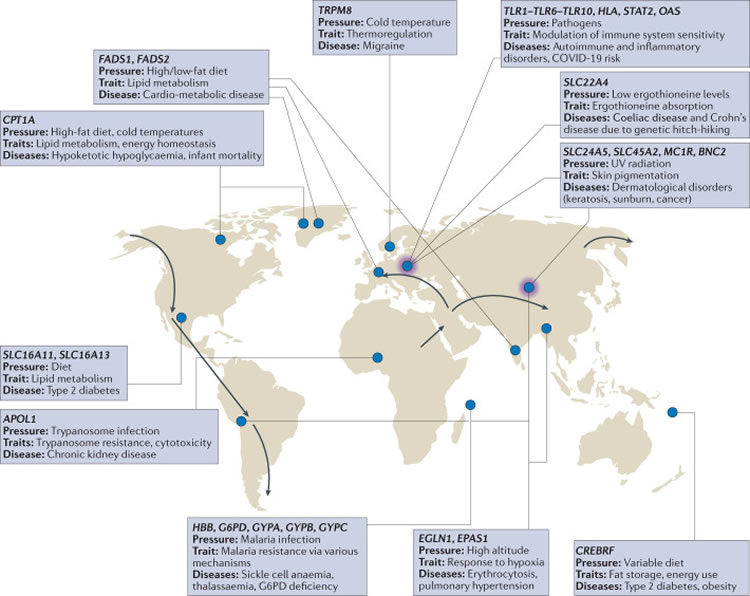
Figure 2 : L'adaptation récente a produit des compromis évolutifs qui mènent à des maladies dans certains environnements.
Voici les gènes représentatifs qui ont connu une évolution adaptative locale au cours des 100 000 dernières années à mesure que les humains se déplaçaient à travers le monde. Nous nous concentrons sur les adaptations qui ont également produit un potentiel de maladie en raison de compromis ou d'inadéquations avec les environnements modernes. Pour chacun, nous listons la pression évolutionniste, le (s) trait (s) influencé (s) et la (les) maladie (s) associée (s). Les régions approximatives où les adaptations se sont produites sont indiquées par des cercles bleus. Les flèches représentent l'expansion des populations humaines et les ombres violettes représentent les événements d'introgression avec des hominines archaïques. Le tableau supplémentaire S1 présente plus de détails et de références.
COVID-19 : maladie à coronavirus 2019 - G6PD :glucose-6-phosphate déshydrogénase - UV : ultraviolet.
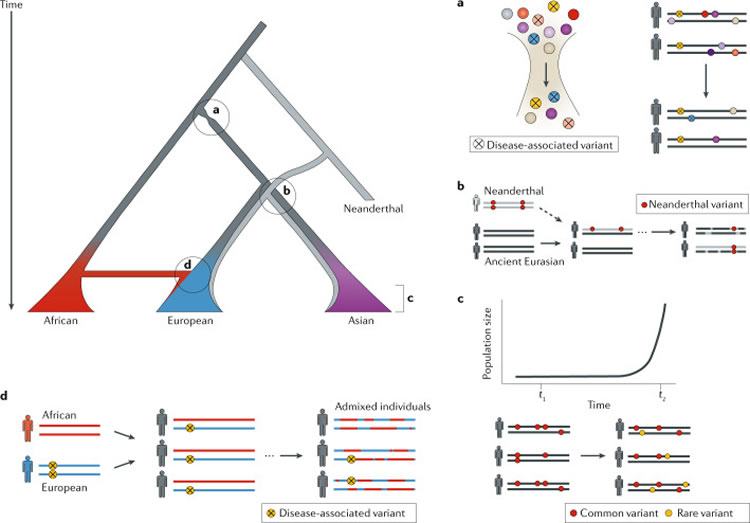
Figure 3 : Effets des événements démographiques récents dans l'histoire humaine sur les mécanismes génétiques sous-jacents à la maladie.
Les migrations humaines anciennes, les événements d'introgression avec d'autres hominidés archaïques et les récentes expansions de population ont tous contribué à l'introduction de variants associés à la maladie humaine.
Schéma de l'histoire de l'évolution humaine, où les branches représentent différentes populations humaines et les largeurs de branches représentent la taille de la population (en haut à gauche). Les étiquettes de lettre font référence aux processus illustrés dans les parties a à d.
a/ Les populations humaines qui migrent hors d'Afrique n'ont conservé qu'un sous-ensemble de la diversité génétique présente dans les populations africaines. Le goulot d'étranglement qui en résulte en dehors de l'Afrique a probablement augmenté la proportion de variants délétères associés à la maladie dans les populations non africaines. Les cercles colorés représentent différents variants génétiques. Les cercles marqués d'un X indiquent des variants délétères associés à la maladie.
b/ Lorsque les humains anatomiquement modernes ont quitté l'Afrique, ils ont rencontré d'autres populations d'hominidés archaïques. Les haplotypes introduits par des événements d'introgression archaïques (illustrés en gris) contenaient des variants dérivés de Néandertal (indiqués par des cercles rouges) associés à un risque accru de maladie dans les populations modernes.
c/ Au cours des 10 000 dernières années, le fardeau des variants associés à des maladies rares (indiquées par des cercles jaunes) a augmenté en raison de la rapide expansion démographique.
d/ Les individus humains modernes avec un mélange dans leur ascendance récente, comme les Afro-Américains, peuvent avoir des différences de risque génétique de maladie, en raison du mélange unique de régions génomiques de chaque individu avec une ascendance africaine et européenne. Par exemple, chacun des trois individus mélangés a les mêmes proportions d'ascendance africaine et européenne, mais tous ne portent pas le variant associé à une maladie plus fréquente en Europe (illustrée par des cercles jaunes). Résumer le risque clinique pour un patient nécessite une vision à plus haute résolution dans la phylogenèse du génome et une meilleure représentation des variations génétiques des diverses populations humaines.
Il y a environ 200 000 ans, les «humains anatomiquement modernes» (AMH) sont apparus pour la première fois en Afrique. Ce groupe avait les principales caractéristiques physiques des groupes humains actuels et présentait des capacités comportementales et cognitives uniques qui ont permis des améliorations rapides dans le développement d'outils, l'art et la culture matérielle. Il y a environ 100 000 ans, des groupes AMH ont commencé à émigrer hors d'Afrique. Les populations ancestrales de tous les Eurasiens modernes ont probablement quitté l'Afrique des dizaines de milliers d'années plus tard [98], mais se sont rapidement propagées à travers l'Eurasie. On pense que des expansions aux Amériques et d'autres goulots d'étranglement se sont produits il y a entre 35 000 et 15 000 ans. Les détails et les incertitudes entourant ces événements d'origine et de migration sont examinés plus en détail ailleurs [98].
Les populations qui subissent des goulots d'étranglement et des effets fondateurs ont une charge mutationelle plus élevée que les autres, en grande partie en raison de leur taille de population effective inférieure réduisant l'efficacité de la sélection [100] (Fig. 3a). Au cours de cette dispersion, les populations humaines migrantes présentaient moins de variations génétiques que celles présentes en Afrique. La réduction de la diversité causée par ce « out-of-Africa » et les goulots d’étranglement qui en résultent ont façonné le paysage génétique de toutes les populations non africaines.
Les AMH n'ont pas vécu isolément après la migration hors d'Afrique. Au lieu de cela, il existe des preuves de multiples mélanges avec d'autres groupes d'hominidés archaïques, à savoir les Néandertaliens et les Denisoviens [101, 102]. Les populations non africaines modernes tirent environ 2% de leur ascendance des Néandertaliens, certaines populations asiatiques ayant une proportion encore plus élevée d'ascendance hominine archaïque (Fig. 3b). Les populations africaines n'ont que très peu d'ascendance néandertalienne et denisovienne, en grande partie par les migration de retour des populations européennes d'ascendance archaïque [103]. Cependant, il existe des preuves de mélange avec d'autres hominines archaïques, encore inconnus, dans les génomes des populations africaines modernes [104, 105, 106].
Suite à leur expansion dans le monde entier, les humains ont connu une croissance explosive au cours des 10 000 dernières années, en particulier dans les populations eurasiennes modernes [107, 108] (Fig. 3c). La croissance de la taille de la population modifie l'architecture génétique des caractères en augmentant l'efficacité de la sélection et en générant beaucoup plus de variants génétiques à basse fréquence. Bien que l'impact des allèles rares ne soit pas complètement compris, ils ont souvent un rôle délétère dans la variation des caractères des populations modernes [109]. Bien qu'il y ait encore débat sur les effets combinés de ces différences démographiques récentes, un consensus se dégage selon lequel elles n'auront probablement que des effets mineurs sur l'efficacité de la sélection et la charge de mutation entre les populations humaines [100, 110, 111, 112, 113, 114]. Néanmoins, les différences substantielles de fréquence des allèles entre populations sont pertinentes pour le risque de maladie [115].
L'exposition des humains à de nouveaux environnements et à d'importants changements de mode de vie, tels que l'agriculture et l'urbanisation, a créé une opportunité d'adaptation [96, 116]. Les déjà multiples séquençages de l'ADN, associés aux progrès statistiques récents, commencent à permettre de relier les adaptations humaines à ces changements environnementaux spécifiques récents [96, 117, 118]. Cependant, ces changements environnementaux rapides ont également créé de nouveaux modèles de maladies complexes. L'inadéquation entre notre aptitude biologique pour les environnements ancestraux et les environnements modernes explique la prévalence de nombreuses maladies courantes, telles que l'obésité, le diabète de type 2 ou les maladies cardiaques, qui découlent de modes de vie sédentaires et d'une mauvaise alimentation. Le modèle de susceptibilité ancestrale propose que les allèles ancestraux qui ont été adaptés aux environnements anciens peuvent, dans les populations modernes, augmenter le risque de maladie [119, 120]. Plusieurs études viennent encore appuyer cette hypothèse [121, 122]. Cependant, soulignant l'importance de l'histoire démographique récente, les modèles de risque pour les allèles ancestraux et dérivés diffèrent dans les populations africaines et européennes, avec des allèles à risque ancestraux à des fréquences plus élevées dans les populations africaines [115].
Implications médicales
Les différentes histoires évolutionnistes des individus et des populations humaines modernes décrites dans la section précédente influencent les risques et fréquences des maladies. Ce qui est peut-être le plus frappant, ce sont les inadéquations et les compromis résultant des récentes adaptations du système immunitaire. Les exemples classiques comprennent les variants génétiques conférant une résistance au paludisme provoquant la drépanocytose et autres anomalies de l’hémoglobine chez les homozygotes [96, 123], ou les variants à prédominance africaine G1 et G2 dans APOL1 protégeant contre la maladie du sommeil (trypanosomiase)' mais conduisant à une maladie rénale chronique chez les individus portant ces génotypes [96] . De même, une variante du CREBRF qui aurait amélioré la survie des personnes en période de famine est désormais liée à l'obésité et au diabète de type 2 [124]. Dans une étude sur d'anciennes populations européennes, un variant du SLC22A4, le transporteur de l'ergothionéine, qui a été sélectionné pour se protéger contre une carence en cet antioxydant est également associé à des problèmes gastro-intestinaux tels que la maladie cœliaque, la colite ulcéreuse et le syndrome du côlon irritable [118] . Le variant responsable n'a atteint une fréquence élevée dans les populations européennes que relativement récemment, et son association à de nouvelles maladies résulte probablement d’une inadéquation avec l'environnement actuel [118]. La possibilité d'une discordance est en outre étayée par la prévalence variable de la maladie cœliaque entre les populations liée à la sélection de plusieurs allèles à risque [82]. En effet, des études récentes suggèrent qu'il existe une relation entre l'ascendance et la réponse immunitaire, les individus d'ascendance africaine ayant des réponses plus fortes. Cela pourrait être le résultat de processus sélectifs en réponse à de nouveaux environnements pour les populations européennes, ou une pression d'agents pathogènes en Afrique conduisant maintenant à plus de cas de maladies inflammatoires et auto-immunes. Mais ce domaine de recherche est très récent il faut davantage de preuves pour établir des conclusions solides [125].
Dans les environnements humains modernes, il existe également un décalage entre les faibles niveaux actuels d'infection parasitaire et le système immunitaire qui a évolué sous une charge parasitaire plus élevée. Cette discordance est supposée contribuer à l'augmentation des maladies inflammatoires et auto-immunes observées de nos jours [34]. Par exemple, des loci associés à dix maladies inflammatoires différentes, y compris la maladie de Crohn et la sclérose en plaques, montrent des preuves de sélection compatibles avec cette hypothèse hygiéniste [126]. De plus, une sélection positive récente sur des variants de la voie de la réponse immunitaire de type 2 a favorisé les allèles de sensibilité à l'asthme [127]. Cela suggère que des processus évolutifs récents peuvent avoir conduit à des réponses immunitaires élevées ou modifiées au détriment d'une sensibilité accrue aux maladies inflammatoires et auto-immunes. Cette approche a de vastes implications cliniques, y compris l'utilisation ciblée d’helminthes et de leurs productions naturelles pour la modulation immunitaire chez les patients atteints d'une maladie inflammatoire chronique [128, 129].
L'introgression archaïque est pertinente pour la médecine moderne car les allèles introduits par ces événements évolutionnistes continuent d'avoir un impact sur les populations modernes même si les lignées d'hominidés archaïques sont éteintes (Fig. 3b). Les hominines archaïques avaient des tailles de population efficaces considérablement plus faibles que les AMH, et donc elles portaient probablement une plus grande fraction de mutations faiblement délétères que les AMH [101]. En conséquence, on suppose que l'introgression néandertalienne a considérablement augmenté la charge génétique des AMH non africaines [130, 131]. Des efforts de séquençage à grande échelle, en combinaison avec l'analyse de biobanques cliniques et des méthodes de calcul améliorées, ont révélé les impacts potentiels de ces introgressions d'ADN sur les génomes humains modernes. Plusieurs études récentes relient les régions de ces introgressions à une gamme de maladies, dont plusieurs phénotypes immunologiques, neuropsychiatriques et dermatologiques [102, 132, 133, 134, 135, 136, 137, 138, 139]. Cela démontre l'impact fonctionnel de ces introgressions de séquences sur le risque de maladie chez les humains non africains d’aujourd'hui. Cependant, certaines de ces associations peuvent être influencées par des allèles liés non néandertaliens. Par exemple, l'introgression a aussi concerné des allèles ancestraux qui avaient été perdus dans les populations eurasiennes modernes avant le métissage (par exemple, lors du goulot d'étranglement du out-of-Africa) [141]. Certains allèles introgressés peuvent avoir initialement atténué les effets néfastes de la migration vers les climats nordiques, des changements alimentaires et de l'introduction de nouveaux pathogènes [117, 142, 143]. Par exemple, des allèles de Néandertal contribuent à la variation de l’immunité innée entre les populations [125, 132, 134, 144] et ont probablement aidé les AMH à s'adapter à de nouveaux virus, en particulier les virus à ARN en Europe [145]. Cependant, en raison des récents changements démographiques et environnementaux, certains allèles de Néandertal auparavant adaptatifs peuvent ne plus offrir les mêmes avantages [146]. Par exemple, il est prouvé qu'un haplotype de Néandertal introgressé augmente le risque de SRAS-CoV-2 [147].
Les médecins, dans leur pratique clinique, ont l’habitude de noter les antécédents familiaux et les origines ethniques et géographiques déclarés par leurs patients ; cependant, ceci ne suffit évidemment pas à saisir l'ascendance évolutionniste complexe de chaque patient. Par exemple, deux individus qui s'identifient comme Afro-Américains peuvent tous deux avoir 15% d'ascendance européenne, mais cette ascendance sera à des locus génomiques différents et proviendra de différentes populations ancestrales européennes et africaines (Fig. 3d). Ainsi, l'un peut être porteur d'un allèle d'ascendance européenne qui augmente une maladie, et pas l'autre. La cartographie de l'ascendance génétique à petite échelle dans les génomes des patients peut améliorer notre capacité à résumer les risques cliniquement pertinents [148], mais de telles approches nécessitent un large échantillonnage parmi les populations et une prise de conscience de la diversité humaine (encadré 4). Il y a un grand besoin d'augmenter l'échantillonnage de divers groupes, car il y a une surreprésentation des populations d'ascendance européenne qui limite l’étude des risques pour les autres populations [149, 150, 151] (Fig. 4). En 2016, 81% des données des GWAS provenaient d'études menées sur des populations européennes [149]. Bien qu'il s'agisse d'une amélioration par rapport aux 96% de 2009, la plupart des populations non européennes manquent encore d’une bonne représentation dans ces études. Le problème est plus extrême pour de nombreux phénotypes ou traits d'intérêt. Par exemple, seulement 1,2% des études d'une enquête auprès de 569 GWAS sur certains phénotypes neurologiques incluaient des personnes d'ascendance africaine [150, 152].
Les biais d'ascendance dans les bases de données génomiques et les GWAS se propagent à d’autres connaissances génétiques utiles en clinique, comme les scores de risque polygénique (PRS) [153, 154] (Fig. 4b). Les PRS peuvent prédire des résultats médicaux à partir des seules données génomiques. Cependant, la perspective évolutionniste suggère que l'architecture génétique des maladies diffère entre les populations en raison des différences démographiques et environnementales évoquées ci-dessus. En effet, de nombreuses PRS ne sont pas généralisables entre populations et sont sujettes à des biais [155, 156]. La hiérarchisation des gènes de maladie mendélienne est également difficile dans les populations sous-représentées. En général, les individus d'ascendance africaine ont beaucoup plus de variants, mais nous en savons moins sur la pathogénicité de ceux de ces variants qui sont absents ou moins fréquents dans les populations européennes [157]. Les patients d'ascendance africaine et asiatique sont actuellement plus susceptibles que ceux d'ascendance européenne d’avoir des résultats de tests génétiques ambigus après le séquençage de leur exome ou de se faire dire qu'ils ont des variants de signification incertaine [158]. La sous-représentation des variants pathogènes d'origine non européenne dans les bases de données [159] couvre une gamme de traits phénotypiques, comme les effets des variants du CYP2D6 sur la réponse aux médicaments [160, 161], l'identification et la classification des risques de cancer du sein [162], et les effets disparates des GWAS pour l'indice de masse corporelle et le diabète de type 2 [163]. Dans une étude sur la cardiomyopathie hypertrophique, les variants bénins chez les Afro-Américains ont été incorrectement classées comme pathogènes sur la base des résultats du GWAS d'une cohorte d'ascendance européenne. L'inclusion d'individus d'origine africaine dans les GWAS initiaux aurait pu éviter ces erreurs [164].
Encadré 4 : Médecine évolutionniste dans la pratique clinique
Les perspectives évolutionnistes doivent encore être intégrées dans la plupart des domaines de la pratique clinique. Les exceptions notables concernent les maladies dans lesquelles les processus évolutionnistes agissent sur des échelles de temps courtes. Par exemple, la connaissance des pressions sélectives intenses sous-jacentes à l'évolution de la résistance aux antibiotiques et antiviraux et à la croissance des tumeurs guide désormais l'application de stratégies thérapeutiques précises [210, 211, 212, 213]. Ces exemples illustrent comment une perspective évolutionniste peut améliorer certains résultats thérapeutiques. Cependant, ils diffèrent de l'objectif principal de cet article consacré aux influences génétiques qui ont agi sur des milliers ou des millions d'années.
Néanmoins, la prise en compte des innovations, des adaptations et des compromis qui ont façonné les populations humaines doit être prise en compte dans l'application clinique de la médecine de précision à des maladies complexes. Par exemple, les scores de risque polygénique (PRS) sont une technologie en plein essor avec un grand potentiel pour stratifier les risques cliniques des individus et permettre des soins préventifs [154, 214], mais ils dépendent fondamentalement des processus évolutionnistes sous-jacents. Les individus ont des antécédents génétiques différents en fonction de leur ascendance, et ces différentes histoires modifient les relations entre les génotypes, les facteurs environnementaux et le risque de maladie (Fig. 4). Dans cette perspective évolutionniste, il ne faut pas s'attendre à ce que les PRS se généralisent à travers les populations et les environnements étant donné les histoires démographiques variées des populations humaines qui façonnent la variation génétique [155, 156, 215]. En effet, le fait de ne pas tenir compte de cette diversité dans l'application des PRS et d'autres méthodes de prédiction fondées sur la génétique peut causer des dommages substantiels et contribuer aux disparités en matière de santé en produisant des diagnostics erronés, des dosages incorrects des médicaments et des prévisions de risque inexactes [149, 150, 151, 158,160, 161, 162, 163,164]. L’approche évolutionniste permet de résoudre ces problèmes. Les PRS doivent être élaborées et évaluées de manière critique sur l'ensemble de la diversité humaine afin de déterminer quand les facteurs génétiques peuvent fournir un profil de risque individuel précis. Ceci est crucial chez les individus de métissage récent, car les profils de risque peuvent varier en fonction des modèles d'ascendance uniques de chaque individu (Fig. 3). Si l'information génétique doit informer les prédictions personnalisées sur le risque de maladie, la prise en compte explicite de l'évolution en quantifiant l'ascendance génétique doit être un élément essentiel de ce processus.
Le développement des PRS fournit une étude de cas opportune et illustrative de la façon dont les perspectives évolutionnistes peuvent passer des contextes de recherche aux applications cliniques. Il met également en évidence les écueils liés à l'ignorance des implications de l'histoire de l'évolution humaine lors de la généralisation des résultats à travers les populations. La mise en place d'une nouvelle technologie (séquençage du génome) a permis de mesurer un signal informatif sur le risque de maladie (variation génétique) mais aussi influencé par l'histoire évolutionniste. Les connaissances acquises au cours de 100 ans de recherche fondamentale en génétique des populations fournissent le contexte de ces nouvelles technologies et la voie à suivre pour garantir que les nouveaux traitements ne soient pas biaisés contre des populations spécifiques.
Au-delà du contexte des analyses et des traitements existants, de nouvelles approches sont nécessaires pour traduire nos connaissances en biologie de l’évolution de la recherche fondamentale à la pertinence clinique. Dans le texte principal, nous avons donné des exemples sur la façon dont les compromis, causés par la concurrence pour les ressources ou la pléiotropie antagoniste, peuvent produire des effets contrastés sur le risque de maladie chez un individu. De même, de nouvelles conditions environnementales, comme un nouveau pathogène, peuvent rapidement créer des inadéquations génétiques dans certaines populations. Nous proposons que l'analyse guidée par les biobanques liées aux dossiers de santé électroniques soit une approche prometteuse pour identifier de nouveaux modèles de maladie diamétrale ou de mésappariements dans les populations de patients. Par exemple, si un gène aux fonctions pléiotropes est ciblé par un traitement, tel qu’un médicament, la connaissance de l’évolution et des fonctions du gène peut suggérer des phénotypes spécifiques pour tester l’occurrence diamétrale dans la biobanque. Étant donné le chevauchement des histoires évolutionnistes des voies moléculaires impliquées dans la plupart des caractères, nous pensons que de nombreux compromis cliniquement pertinents restent à découvrir.
Conclusions et perspectives
Toutes les maladies ont des histoires évolutionnistes, et les signatures de ces histoires sont archivées dans nos génomes. Les progrès récents de la génomique nous permettent de lire ces histoires avec une précision, une résolution et une profondeur élevées. Les aperçus de la génomique évolutionniste révèlent qu'il n'y a pas une seule réponse à la question de savoir pourquoi nous tombons malades. Les maladies affectent plutôt des patchworks d'anciens systèmes biologiques qui ont évolué au cours des millénaires, et bien que les systèmes impliqués soient anciens, la variation pertinente pour les maladies humaines est récente. De plus, les approches de la génomique évolutionniste ont le pouvoir d'identifier les mécanismes, voies et réseaux potentiels et de suggérer des cibles cliniques. Dans ce contexte, nous soutenons qu'une perspective évolutionniste peut aider à la mise en œuvre de la médecine de précision à l'ère du séquençage et de l'édition du génome [165] (encadré 4).
La combinaison de la connaissance des événements évolutionnistes dans la lignée humaine avec les résultats d'études génomiques récentes fournit un cadre explicatif au-delà des descriptions de risque de maladie ou d'association. Par exemple, une analyse récente de l'incidence plus élevée du cancer de la prostate chez les hommes d'ascendance africaine a non seulement découvert un ensemble de variants génétiques associés à un risque accru, mais a également utilisé des mesures de sélection pour proposer l’explication de l'auto-stop génétique [166]. Les haplotypes ayant des effets protecteurs contre le cancer de la prostate peuvent avoir été plus fréquents dans les populations non africaines en raison de la sélection sur des variants proches liés à la pigmentation de la peau (Fig. 4c). Ainsi, les perspectives évolutionnistes aident non seulement à répondre à la question de savoir comment nous tombons malades, mais aussi pourquoi nous tombons malades.
À mesure que les informations génétiques disponibles auprès de diverses populations augmentent, nous pouvons cartographier spécifiquement la génétique des caractères dans différentes populations et définir plus précisément le risque de maladie sur une base individuelle [167, 168]. Cependant, soulignons que les facteurs environnementaux et sociaux sont des déterminants majeurs du risque de maladie, souvent plus que la génétique, et doivent donc être priorisés. L'étude de diverses populations humaines fournira une puissance supplémentaire pour découvrir les loci associés aux traits et comprendre l'architecture génétique à travers différentes expositions environnementales et histoires évolutionnistes [150, 169]. Par exemple, une GWAS à petite taille d'échantillon dans une population inuite du Groenland a trouvé une variante dans une enzyme d'acide gras qui affecte la taille à la fois dans cette population et dans les populations européennes [170]. La GWAS précédente a probablement manqué cette variante en raison de sa faible fréquence dans les populations européennes (0,017 contre 0,98 chez les Inuits) ; néanmoins, il a un effet plus fort sur la taille que les autres variants précédemment identifiés [170]. De même, une étude récente de la taille chez 3000 Péruviens a identifié une autre variante ayant une influence encore plus grande sur la taille [171]. La croissance de grandes biobanques d’ADN dans lesquelles des centaines de milliers de patients sont liés à des échantillons d’ADN représente une ressource inexploitée substantielle pour la médecine évolutionniste [5, 86, 87]. Ces données permettent de tester les effets fonctionnels des variants génétiques sur divers caractères à un coût supplémentaire minime. Le passage d'une GWAS à une seule ascendance à une GWAS trans-ethnique ou multiethnique aura le double avantage d’un plus grand échantillon, et d’une plus grande diversité signaux [172, 173, 174, 175].
Bien que les hypothèses évolutionnistes soient tacites dans la pratique médicale, les antécédents familiaux autodéclarés restaient jusqu'à récemment la meilleure représentation de l'empreinte de notre ascendance sur notre risque de maladie. Cependant, une histoire familiale ne peut pas saisir entièrement l'histoire évolutionniste et démographique complexe de chaque individu. Les nouvelles technologies permettent désormais de recueillir et d’interpréter l’histoire familiale d’un individu sous une forme beaucoup plus longue et complémentaire : son génome. Les nouvelles données et méthodes augmentent considérablement la résolution et la profondeur avec lesquelles ces histoires peuvent être quantifiées, offrant des opportunités pour la pratique médicale.
Tableau supplémentaire S1
| Gènes | Pression environnementale | Trait | Maladie | ref |
| FADS1/2 | Régime riche ou pauvre en graisses | Métabolisme lipidique | Maladies cardiovasculaires | 1,2,3,4,5,6 |
| CPT1A | Régime riche en graisses froides températures |
métabolisme lipidique Homéostase énergétique Taille |
Hypoglycémie hypocétosique Mortalité infantile élevée |
2,4,7 |
| SLC22A4 | Bas niveaux d'ergothionéine | Absorption d'ergothionéine | Maladie coeliaque Rectocolite hémorragique Sybdrome du côlon irritable |
2,8,9 |
| CREBRF | Famine | Stockage des graisses Utilisation de l'anergie |
Diabète de type 2 Obésité |
1,2,10 |
| HBB, G6PD, GYPA, GYPB, GYPC |
Paludisme | Résistance au paludisme | Drépanocytose Thalassémie Déficit en G6PD |
1,2 |
| APOL1 | Trypanosomiase (Maladie du sommeil) |
Résistance au trypanosome Cytotoxicité |
Maladie rénale chronique | 1,2,11 |
| TRPM8 | Froides températures | Thermorégulation | Migraine | 1,2,12 |
| SLC24A5 SLC45A2 MC1R, BNC2 |
Radiations UV | Pigmentation cutanée | troubles dermatologiques Kératose, coups de soleil |
2,9,13,14 |
| TLR6,TLR1 TLR10, HLA STAT2, OAS |
Pathogène | Sensibilité immunitaire | Maladies auto-immunes Maladies inflammatoires |
15,2 |
| SLC16A11 SLC16A13 |
Régime | Métabolisme lipidique | Diabète de type 2 | 2,21 |
| EGLN1 EPAS1 |
Haute altitude | Réponse à l'hypoxie | Érythrocytose Hypertension pulmonaire |
22,25 |
Traduction : Luc Perino
Bibliographie
Benton ML, Abraham A, LaBella AL, Abbot P, Rokas A, Capra JA.
The influence of evolutionary history on human health and disease
Nat Rev Genet. 2021;1-15.
DOI : 10.1038/s41576-020-00305-9
Références numérotées
1. Alföldi J, Lindblad-Toh K. Comparative genomics as a tool to understand evolution and disease. Genome Res. 2013;23:1063–1068.
2. Meadows JRS, Lindblad-Toh K. Dissecting evolution and disease using comparative vertebrate genomics. Nat. Rev. Genet. 2017;18:624–636.
3. Shendure J, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550:345–353.
4. Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443.
5. Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209.
6. Dewey FE, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814.
7. McCarty CA, et al. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics. 2011;4:13.
8. Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genet. 2018;19:110–124.
9. Orlando L, Gilbert MTP, Willerslev E. Reconstructing ancient genomes and epigenomes. Nat. Rev. Genet. 2015;16:395–408.
10. Skoglund P, Mathieson I. Ancient genomics of modern humans: the first decade. Annu. Rev. Genomics Hum. Genet. 2018;19:381–404.
11. Ramamoorthy A, Yee SW, Karnes J. Unveiling the genetic architecture of human disease for precision medicine. Clin. Transl. Sci. 2019;12:3–5.
12. Stearns, S. C. & Medzhitov, R. Evolutionary Medicine (Sinauer Associates, 2016). This foundational textbook provides an introduction to the field of evolutionary medicine.
13. Carroll SP, et al. Applying evolutionary biology to address global challenges. Science. 2014;346:1245993.
14. Young MJ, Copeland WC. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016;38:52–62.
15. Muñoz S, Méndez J. DNA replication stress: from molecular mechanisms to human disease. Chromosoma. 2017;126:1–15.
16. Ackermann M, Chao L, Bergstrom CT, Doebeli M. On the evolutionary origin of aging. Aging Cell. 2007;6:235–244.
17. Flatt T, Partridge L. Horizons in the evolution of aging. BMC Biol. 2018;16:93.
18. Rokas A. The origins of multicellularity and the early history of the genetic toolkit for animal development. Annu. Rev. Genet. 2008;42:235–251.
19. Albuquerque TAF, Drummond do Val L, Doherty A, de Magalhães JP. From humans to hydra: patterns of cancer across the tree of life. Biol. Rev. 2018;93:1715–1734.
20. Crespi B, Summers K. Evolutionary biology of cancer. Trends Ecol. Evol. 2005;20:545–552.
21. Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761.
22. Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat. Rev. Cancer. 2004;4:197–205.
23. Domazet-Lošo T, Tautz D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010;8:66.
24. Thomas F, et al. Applying ecological and evolutionary theory to cancer: a long and winding road. Evol. Appl. 2013;6:1–10.
25. Vogelstein B, et al. Cancer genome landscapes. Science. 2013;340:1546–1558.
26. Aktipis CA, Nesse RM. Evolutionary foundations for cancer biology. Evol. Appl. 2013;6:144–159.
27. Gerlinger M, et al. Cancer: evolution within a lifetime. Annu. Rev. Genet. 2014;48:215–236.
28. Enriquez-Navas PM, Wojtkowiak JW, Gatenby RA. Application of evolutionary principles to cancer therapy. Cancer Res. 2015;75:4675–4680.
29. Gerstung M, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–128.
30. Hibino T, et al. The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 2006;300:349–365.
31. Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–329.
32. Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat. Rev. Genet. 2010;11:47–59.
33. Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351:1083–1087.
34. Dunne DW, Cooke A. A worm’s eye view of the immune system: consequences for evolution of human autoimmune disease. Nat. Rev. Immunol. 2005;5:420–426.
35. Shubin N, Tabin C, Carroll S. Deep homology and the origins of evolutionary novelty. Nature. 2009;457:818–823.
36. Shubin, N. Your Inner Fish: A Journey into the 3.5-billion-year History of the Human Body (Knopf Doubleday, 2008).
37. Guernsey MW, Chuong EB, Cornelis G, Renfree MB, Baker JC. Molecular conservation of marsupial and eutherian placentation and lactation. eLife. 2017;6:e27450.
38. Abbot P, Capra JA. What is a placental mammal anyway?: many developmental functions in marsupials and eutherian mammals are accomplished by different tissues, but similar genes. eLife. 2017;6:e30994.
39. Katz PS. ‘Model organisms’ in the light of evolution. Curr. Biol. 2016;26:R649–R650.
40. Bolker JA. Selection of models: evolution and the choice of species for translational research. Brain Behav. Evol. 2019;93:82–91.
41. Bart van der Worp H, et al. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7:1–8.
42. Mak IWY, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014;6:114–118.
43. Caulin AF, Maley CC. Peto’s paradox: evolution’s prescription for cancer prevention. Trends Ecol. Evol. 2011;26:175–182.
44. Tollis M, et al. Return to the sea, get huge, beat cancer: an analysis of cetacean genomes including an assembly for the humpback whale (Megaptera novaeangliae) Mol. Biol. Evol. 2019;36:1746–1763.
45. Sulak M, et al. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife. 2016;5:e11994.
46. Vazquez JM, Sulak M, Chigurupati S, Lynch VJ. A zombie LIF gene in elephants is upregulated by TP53 to induce apoptosis in response to DNA damage. Cell Rep. 2018;24:1765–1776.
47. O’Bleness M, Searles VB, Varki A, Gagneux P, Sikela JM. Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 2012;13:853–866.
48. Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. Genetic evidence for complex speciation of humans and chimpanzees. Nature. 2006;441:1103–1108.
49. Crespi BJ, Go MC. Diametrical diseases reflect evolutionary-genetic tradeoffs: evidence from psychiatry, neurology, rheumatology, oncology and immunology. Evol. Med. Public Health. 2015;2015:216–253.
50. Stearns SC. Trade-offs in life-history evolution. Funct. Ecol. 1989;3:259.
51. Ricklefs RE, Wikelski M. The physiology/life-history nexus. Trends Ecol. Evol. 2002;17:462–468.
52. Zera AJ, Harshman LG. The physiology of life history trade-offs in animals. Annu. Rev. Ecol. Syst. 2001;32:95–126.
53. Brady SP, et al. Understanding maladaptation by uniting ecological and evolutionary perspectives. Am. Nat. 2019;194:495–515.
54. Ellison PT. Evolutionary tradeoffs. Evol. Med. Public Health. 2014;2014:93.
55. Al-Nbaheen M, et al. Human stromal (mesenchymal) stem cells from bone marrow, adipose tissue and skin exhibit differences in molecular phenotype and differentiation potential. Stem Cell Rev. Rep. 2013;9:32–43.
56. Jacqueline C, et al. Cancer: a disease at the crossroads of trade-offs. Evol. Appl. 2017;10:215–225.
57. Marques-Bonet T, Eichler EE. The evolution of human segmental duplications and the core duplicon hypothesis. Cold Spring Harb. Symposia Quant. Biol. 2009;74:355–362.
58. Dennis MY, Eichler EE. Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 2016;41:44–52.
59. Fiddes IT, et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell. 2018;173:1356–1369.e22.
60. Suzuki IK, et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through Delta/Notch regulation. Cell. 2018;173:1370–1384.e16.
61. Guerrier S, et al. The F-BAR domain of srGAP2 induces membrane protrusions required for neuronal migration and morphogenesis. Cell. 2009;138:990–1004.
62. Dennis MY, et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922.
63. Florio M, et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science. 2015;347:1465–1470.
64. Antonacci F, et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat. Genet. 2014;46:1293–1302.
65. Sikela JM, Searles Quick VB. Genomic trade-offs: are autism and schizophrenia the steep price of the human brain? Hum. Genet. 2018;137:1–13.
66. Srinivasan S, et al. Genetic markers of human evolution are enriched in schizophrenia. Biol. Psychiatry. 2016;80:284–292.
67. Polimanti R, Gelernter J. Widespread signatures of positive selection in common risk alleles associated to autism spectrum disorder. PLoS Genet. 2017;13:1–14.
68. Xu K, Schadt EE, Pollard KS, Roussos P, Dudley JT. Genomic and network patterns of schizophrenia genetic variation in human evolutionary accelerated regions. Mol. Biol. Evol. 2015;32:1148–1160.
69. Doan RN, et al. Mutations in human accelerated regions disrupt cognition and social behavior. Cell. 2016;167:341–354.e12.
70. Sharp PM, Hahn BH. The evolution of HIV-1 and the origin of AIDS. Phil. Trans. R. Soc. B. 2010;365:2487–2494.
71. Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proc. Natl Acad. Sci. USA. 2006;103:7765–7770.
72. Soto PC, Karris MY, Spina CA, Richman DD, Varki A. Cell-intrinsic mechanism involving Siglec-5 associated with divergent outcomes of HIV-1 infection in human and chimpanzee CD4 T cells. J. Mol. Med. 2013;91:261–270.
73. Arora G, Polavarapu N, McDonald JF. Did natural selection for increased cognitive ability in humans lead to an elevated risk of cancer? Med. Hypotheses. 2009;73:453–456.
74. Varki NM, Varki A. On the apparent rarity of epithelial cancers in captive chimpanzees. Phil. Trans. R. Soc. B. 2015;370:20140225.
75. Varki A. Loss of N-glycolylneuraminic acid in humans: mechanisms, consequences, and implications for hominid evolution. Yearb. Phys. Anthropol. 2001;44:54–69.
76. Chou HH, et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo–Pan divergence. Proc. Natl Acad. Sci. USA. 1998;95:11751–11756.
77. Irie A, Koyamat S, Kozutsumi Y, Kawasaki T, Suzuki A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 1998;273:15866–15871.
78. Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A. Evolution of human–chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc. Natl Acad. Sci. USA. 2005;102:12819–12824.
79. Varki A, Gagneux P. Human-specific evolution of sialic acid targets: explaining the malignant malaria mystery? Proc. Natl Acad. Sci. USA. 2009;106:14739–14740.
80. Deng L, et al. Host adaptation of a bacterial toxin from the human pathogen Salmonella typhi. Cell. 2014;159:1290–1299.
81. Varki A. Uniquely human evolution of sialic acid genetics and biology. Proc. Natl Acad. Sci. USA. 2010;107:8939–8946.
82. Quach H, Quintana-Murci L. Living in an adaptive world: genomic dissection of the genus Homo and its immune response. J. Exp. Med. 2017;214:877–894.
83. Quintana-Murci L. Human immunology through the lens of evolutionary genetics. Cell. 2019;177:184–199.
84. Seluanov A, Gladyshev VN, Vijg J, Gorbunova V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer. 2018;18:433–441.
85. He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–1011.
86. Coppola L, et al. Biobanking in health care: evolution and future directions. J. Transl. Med. 2019;17:172.
87. Bowton E, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci. Transl. Med. 2014;6:234cm3.
88. Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat. Rev. Genet. 2016;17:129–145.
89. van Manen D, et al. Genome-wide association scan in HIV-1-infected individuals identifying variants influencing disease course. PLoS ONE. 2011;6:e22208.
90. Sáez-Cirión A, Pancino G. HIV controllers: a genetically determined or inducible phenotype? Immunol. Rev. 2013;254:281–294.
91. McLaren PJ, et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl Acad. Sci. USA. 2015;112:14658–14663.
92. Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science. 1996;272:67–74.
93. Klepiela P, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–774.
94. Martin MP, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat. Genet. 2007;39:733–740.
95. De Groot NG, et al. AIDS-protective HLA-B*27/B*57 and chimpanzee MHC class I molecules target analogous conserved areas of HIV-1/SIVcpz. Proc. Natl Acad. Sci. USA. 2010;107:15175–15180.
96. Fan S, Hansen MEB, Lo Y, Tishkoff SA. Going global by adapting local: a review of recent human adaptation. Science. 2016;354:54–59.
97. Marciniak S, Perry GH. Harnessing ancient genomes to study the history of human adaptation. Nat. Rev. Genet. 2017;18:659–674.
98. Nielsen R, et al. Tracing the peopling of the world through genomics. Nature. 2017;541:302–310.
99. Azevedo L, Serrano C, Amorim A, Cooper DN. Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response. Hum. Genomics. 2015;9:21.
100. Gravel S. When is selection effective? Genetics. 2016;203:451–462.
101. Prüfer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49.
102. Vernot B, Akey JM. Resurrecting surviving Neandertal linages from modern human genomes. Science. 2014;343:1017–1021.
103. Chen L, Wolf AB, Fu W, Li L, Akey JM. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell. 2020;180:677–687.e16.
104. Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc. Natl Acad. Sci. USA. 2011;108:15123–15128.
105. Hsieh PH, et al. Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies. Genome Res. 2016;26:291–300.
106. Lachance J, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457–469.
107. Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743.
108. Fu W, Akey JM. Selection and adaptation in the human genome. Annu. Rev. Genom. Hum. Genet. 2013;14:467–489.
109. Uricchio LH, Zaitlen NA, Ye CJ, Witte JS, Hernandez RD. Selection and explosive growth alter genetic architecture and hamper the detection of causal rare variants. Genome Res. 2016;26:863–873.
110. Fu Q, et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 2013;23:553–559.
111. Do R, et al. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet. 2015;47:126–131.
112. Simons YB, Turchin MC, Pritchard JK, Sella G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 2014;46:220–224.
113. Simons YB, Sella G. The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives. Curr. Opin. Genet. Dev. 2016;41:150–158.
114. Sella G, Barton NH. Thinking about the evolution of complex traits in the era of genome-wide association studies. Annu. Rev. Genomics Hum. Genet. 2019;20:461–493.
115. Kim MS, Patel KP, Teng AK, Berens AJ, Lachance J. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018;19:179.
116. Rees JS, Castellano S, Andrés AM. The genomics of human local adaptation. Trends Genet. 2020;36:415–428.
117. Huerta-Sanchez E, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512:194–197.
118. Mathieson I, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503.
119. Di Rienzo A, Hudson RR. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 2005;21:596–601.
120. Manus MB. Evolutionary mismatch. Evol. Med. Public Health. 2018;2018:190–191.
121. Gibson G. Decanalization and the origin of complex disease. Nat. Rev. Genet. 2009;10:134–140.
122. Lachance J. Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations. BMC Med. Genomics. 2010;3:57.
123. Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 2005;77:171–192.
124. Minster RL, et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 2016;48:1049–1054.
125. Nédélec Y, et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell. 2016;167:657–669.e21.
126. Raj T, et al. Common risk alleles for inflammatory diseases are targets of recent positive selection. Am. J. Hum. Genet. 2013;92:517–529.
127. Barber MF, Lee EM, Griffin H, Elde NC. Rapid evolution of primate type 2 immune response factors linked to asthma susceptibility. Genome Biol. Evol. 2017;9:1757–1765.
128. Smallwood TB, et al. Helminth immunomodulation in autoimmune disease. Front. Immunol. 2017;8:453.
129. Sobotková K, et al. Helminth therapy—from the parasite perspective. Trends Parasitol. 2019;35:501–515.
130. Harris K, Nielsen R. The genetic cost of neanderthal introgression. Genetics. 2016;203:881–891.
131. Juric I, Aeschbacher S, Coop G. The strength of selection against Neanderthal introgression. PLoS Genet. 2016;12:1–25.
132. Dannemann M, Andrés AM, Kelso J. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 2016;98:22–33.
133. Simonti CN. The phenotype legacy of admixture between modern humans and Neandertals. Science. 2016;351:737–742.
134. Quach H, et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell. 2016;167:643–656.e17.
135. Dannemann M, Kelso J. The contribution of neanderthals to phenotypic variation in modern humans. Am. J. Hum. Genet. 2017;101:578–589.
136. Sankararaman S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–357.
137. Sankararaman S, Mallick S, Patterson N, Reich D. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 2016;26:1241–1247.
138. McCoy RC, Wakefield J, Akey JM. Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell. 2017;168:916–927.e12.
139. McArthur E, Rinker DC, Capra JA. Quantifying the contribution of Neanderthal introgression to the heritability of complex traits. bioRxiv. 2020 doi: 10.1101/2020.06.08.140087. CrossRef
140. Skov L, et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature. 2020;582:78–83.
141. Rinker DC, et al. Neanderthal introgression reintroduced functional ancestral alleles lost in Eurasian populations. Nat. Ecol. Evol. 2020;4:1332–1341.
142. Racimo F, Marnetto D, Huerta-Sánchez E. Signatures of archaic adaptive introgression in present-day human populations. Mol. Biol. Evol. 2017;34:296–317.
143. Bigham AW, Lee FS. Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 2014;28:2189–2204.
144. Deschamps M, et al. Genomic signatures of selective pressures and introgression from Archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 2016;98:5–21.
145. Enard D, Petrov DA. Evidence that RNA viruses drove adaptive introgression between Neanderthals and modern humans. Cell. 2018;175:360–371.e13.
146. Gittelman RM, et al. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol. 2016;26:3375–3382.
147. Zeberg H, Pääbo S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature. 2020;587:610–612.
148. Khera AV, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018;50:1219–1224.
149. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538:161–164.
150. Kelly DE, Hansen MEB, Tishkoff SA. Global variation in gene expression and the value of diverse sampling. Curr. Opin. Syst. Biol. 2017;1:102–108.
151. Hindorff LA, et al. Prioritizing diversity in human genomics research. Nat. Rev. Genet. 2018;19:175–185.
152. Quansah E, McGregor NW. Towards diversity in genomics: the emergence of neurogenomics in Africa? Genomics. 2018;110:1–9.
153. Mostafavi H, et al. Variable prediction accuracy of polygenic scores within an ancestry group. eLife. 2020;9:e48376.
154. Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018;19:581–590.
155. Martin AR, et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 2017;100:635–649.
156. Martin AR, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591.
157. Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177:26–31.
158. Petrovski S, Goldstein DB. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 2016;17:157.
159. Kessler MD, et al. Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry. Nat. Commun. 2016;7:12521.
160. Maisano Delser P, Fuselli S. Human loci involved in drug biotransformation: worldwide genetic variation, population structure, and pharmacogenetic implications. Hum. Genet. 2013;132:563–577.
161. Madian AG, Wheeler HE, Jones RB, Dolan ME. Relating human genetic variation to variation in drug responses. Trends Genet. 2012;28:487–495.
162. Huo D, et al. Comparison of breast cancer molecular features and survival by African and European ancestry in The Cancer Genome Atlas. JAMA Oncol. 2017;3:1654–1662.
163. Carlson CS, et al. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLoS Biol. 2013;11:e1001661.
164. Manrai AK, et al. Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 2016;375:655–665.
165. Lynch M. Mutation and human exceptionalism: our future genetic load. Genetics. 2016;202:869–875.
166. Lachance J, et al. Genetic hitchhiking and population bottlenecks contribute to prostate cancer disparities in men of African descent. Cancer Res. 2018;78:2432–2443.
167. Márquez-Luna C, et al. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017;41:811–823.
168. Shi H, et al. Localizing components of shared transethnic genetic architecture of complex traits from GWAS summary data. Am. J. Hum. Genet. 2020;106:805–817.
169. Crawford NG, et al. Loci associated with skin pigmentation identified in African populations. Science. 2017;358:eaan8433.
170. Fumagalli M, et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science. 2015;349:1343–1347.
171. Asgari S, et al. A positively selected FBN1 missense variant reduces height in Peruvian individuals. Nature. 2020;582:234–239.
172. Li YR, Keating BJ. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 2014;6:1–14.
173. Cook JP, Morris AP. Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur. J. Hum. Genet. 2016;24:1175–1180.
174. Mägi R, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet. 2017;26:3639–3650.
175. Fernández-Rhodes L, et al. Trans-ethnic fine-mapping of genetic loci for body mass index in the diverse ancestral populations of the Population Architecture using Genomics and Epidemiology (PAGE) study reveals evidence for multiple signals at established loci. Hum. Genet. 2017;136:771–800.
176. Hiby SE, et al. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J. Exp. Med. 2004;200:957–965.
177. Yang J, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015;47:1114–1120.
178. Corbett S, Courtiol A, Lummaa V, Moorad J, Stearns S. The transition to modernity and chronic disease: mismatch and natural selection. Nat. Rev. Genet. 2018;19:419–430.
179. Rodríguez JA, et al. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat. Ecol. Evol. 2017;1:1–5.
180. Byars SG, et al. Genetic loci associated with coronary artery disease harbor evidence of selection and antagonistic pleiotropy. PLoS Genet. 2017;13:e1006328.
181. Gluckman PD, Hanson MA. Changing times: the evolution of puberty. Mol. Cell. Endocrinol. 2006;254–255:26–31.
182. Arnold AJ, Fristrup K. The theory of evolution by natural selection: a hierarchical expansion. Paleobiology. 1982;8:113–129.
183. Futuyma DJ. Evolutionary constraint and ecological consequences. Evolution. 2010;64:1865–1884.
184. Gluckman, P. & Hanson, M. Developmental Origins of Health and Disease (Cambridge Univ. Press, 2006).
185. Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 2010;11:17–30.
186. Fay JC. Disease consequences of human adaptation. Appl. Transl. Genomics. 2013;2:42–47.
187. Frank SA, Crespi BJ. Pathology from evolutionary conflict, with a theory of X chromosome versus autosome conflict over sexually antagonistic traits. Proc. Natl Acad. Sci. USA. 2011;108:10886–10893.
188. Abbot P, Rokas A. Mammalian pregnancy. Curr. Biol. 2017;27:R127–R128.
189. Aungst, H. et al. in Power, M. & Schulkin J. in Integrating Evolutionary Biology into Medical Education: For Maternal and Child Healthcare Students, Clinicians, and Scientists Ch. 5 (eds Schulkin, J. & and Power, M.) 91–118 (Oxford Univ. Press, 2019).
190. Redman CWG, Sargent IL. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010;63:534–543.
191. Than NG, et al. Integrated systems biology approach identifies novel maternal and placental pathways of preeclampsia. Front. Immunol. 2018;9:1661.
192. Bergmann A, et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J. Cell. Mol. Med. 2010;14:1857–1867.
193. Turanov AA, et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 2018;36:1164–1173.
194. Robertson SA. Preventing preeclampsia by silencing soluble Flt-1? N. Engl. J. Med. 2019;380:1080–1082.
195. Moorjani P, Amorim CEG, Arndt PF, Przeworski M. Variation in the molecular clock of primates. Proc. Natl Acad. Sci. USA. 2016;113:10607–10612.
196. Marinić M, Lynch VJ. Relaxed constraint and functional divergence of the progesterone receptor (PGR) in the human stem-lineage. PLoS Genet. 2020;16:e1008666.
197. Zeberg H, Kelso J, Pääbo S. The Neandertal progesterone receptor. Mol. Biol. Evol. 2020;37:2655–2660.
198. Clark AG, et al. Positive selection in the human genome inferred from human–chimp–mouse orthologous gene alignments. Cold Spring Harb. Symp. Quant. Biol. 2003;68:479–486.
199. Chen C, et al. The human progesterone receptor shows evidence of adaptive evolution associated with its ability to act as a transcription factor. Mol. Phylogenet. Evol. 2008;47:637–649.
200. LaBella AL, et al. Accounting for diverse evolutionary forces reveals mosaic patterns of selection on human preterm birth loci. Nat. Commun. 2020;11:3731.
201. Schmidt A, Morales-Prieto DM, Pastuschek J, Fröhlich K, Markert UR. Only humans have human placentas: molecular differences between mice and humans. J. Reprod. Immunol. 2015;108:65–71.
202. Hammer A. Immunological regulation of trophoblast invasion. J. Reprod. Immunol. 2011;90:21–28.
203. Erlebacher A. Immunology of the maternal–fetal interface. Annu. Rev. Immunol. 2013;31:387–411.
204. Robinson DP, Klein SL. Pregnancy and pregnancy-associated hormones alter immune responses and disease pathogenesis. Hormones Behav. 2012;62:263–271.
205. Kourtis AP, Read JS, Jamieson DJ. Pregnancy and infection. N. Engl. J. Med. 2014;370:2211–2218.
206. Hiby SE, et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J. Clin. Invest. 2010;120:4102–4110.
207. Moffett A, Chazara O, Colucci F, Johnson MH. Variation of maternal KIR and fetal HLA-C genes in reproductive failure: too early for clinical intervention. Reprod. Biomed. Online. 2016;33:763–769.
208. Moon JM, Capra JA, Abbot P, Rokas A. Immune regulation in eutherian pregnancy: live birth coevolved with novel immune genes and gene regulation. BioEssays. 2019;41:1900072.
209. Muehlenbachs A, Fried M, Lachowitzer J, Mutabingwa TK, Duffy PE. Natural selection of FLT1 alleles and their association with malaria resistance in utero. Proc. Natl Acad. Sci. USA. 2008;105:14488–14491.
210. Andersson DI, et al. Antibiotic resistance: turning evolutionary principles into clinical reality. FEMS Microbiol. Rev. 2020;44:171–188.
211. Enriquez-Navas PM, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016;8:327ra24.
212. Gatenby RA, Brown JS. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020;17:675–686.
213. Fairlamb AH, Gow NAR, Matthews KR, Waters AP. Drug resistance in eukaryotic microorganisms. Nat. Microbiol. 2016;1:16092.
214. Li R, Chen Y, Ritchie MD, Moore JH. Electronic health records and polygenic risk scores for predicting disease risk. Nat. Rev. Genet. 2020;21:493–502.
215. Duncan L, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019;10:3328.
Références du tableau supplémentaire S1
1. Fan, S., Hansen, M. E. B., Lo, Y. & Tishkoff, S. A. Going global by adapting local: A review of recent human adaptation. Science 354, 54–59 (2016).
2. Rees, J. S., Castellano, S. & Andrés, A. M. The Genomics of Human Local Adaptation. Trends in Genetics (2020). doi:10.1016/j.tig.2020.03.006
3. Mathieson, S. & Mathieson, I. FADS1 and the timing of human adaptation to agriculture. Mol. Biol. Evol. (2018). doi:10.1093/molbev/msy180
4. Fumagalli, M. et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349, 1343–7 (2015).
5. Kothapalli, K. S. D. et al. Positive Selection on a Regulatory Insertion-Deletion Polymorphism in FADS2 Influences Apparent Endogenous Synthesis of Arachidonic Acid. Mol. Biol. Evol. (2016). doi:10.1093/molbev/msw049
6. Buckley, M. T. et al. Selection in Europeans on fatty acid desaturases associated with dietary changes. Mol. Biol. Evol. (2017). doi:10.1093/molbev/msx103
7. Clemente, F. J. et al. A selective sweep on a deleterious mutation in CPT1A in Arctic populations. Am. J. Hum. Genet. (2014). doi:10.1016/j.ajhg.2014.09.016
8. Wang, M. H. et al. Contribution of higher risk genes and European admixture to Crohn’s disease in African Americans. Inflamm. Bowel Dis. (2012). doi:10.1002/ibd.22931
9. Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015).
10. Minster, R. L. et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. (2016). doi:10.1038/ng.3620
11. Genovese, G. et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010). doi:10.1126/science.1193032
12. Key, F. M. et al. Human local adaptation of the TRPM8 cold receptor along a latitudinal cline. PLoS Genet. 1–30 (2018). doi:10.1371/journal.pgen.1007298
13. Sturm, R. A. & Duffy, D. L. Human pigmentation genes under environmental selection. Genome Biology (2012). doi:10.1186/gb-2012-13-9-248
14. Dannemann, M. & Kelso, J. The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. Am. J. Hum. Genet. 101, 578–589 (2017).
15. Nédélec, Y. et al. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell (2016). doi:10.1016/j.cell.2016.09.025
16. Quach, H. et al. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell (2016). doi:10.1016/j.cell.2016.09.024
17. Deschamps, M. et al. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am. J. Hum. Genet. 98, 5–21 (2016).
18. Enard, D. & Petrov, D. A. Evidence that RNA Viruses Drove Adaptive Introgression between Neanderthals and Modern Humans. Cell 175, 360-371.e13 (2018).
19. Gittelman, R. M. et al. Archaic Hominin Admixture Facilitated Adaptation to Out-of-Africa Environments. Curr. Biol. 26, 3375–3382 (2016).
20. Zeberg, H. & Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature (2020). doi:10.1038/s41586-020-2818-3
21. Williams Amy, A. L. et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature (2014). doi:10.1038/nature12828
22. Huerta-Sanchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
23. Bigham, A. W. & Lee, F. S. Human high-altitude adaptation: Forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204 (2014).
24. Bigham, A. W. et al. Andean and Tibetan patterns of adaptation to high altitude. Am. J. Hum. Biol. (2013). doi:10.1002/ajhb.22358
25. Crawford, J. E. et al. Natural Selection on Genes Related to Cardiovascular Health in High-Altitude Adapted Andeans. Am. J. Hum. Genet. (2017). doi:10.1016/j.ajhg.2017.09.023
Et pour aller plus loin
Profil de nos 5000 abonnés
| Par catégorie professionnelle | |
| Médecins | 27% |
| Professions de santé | 33% |
| Sciences de la vie et de la terre | 8% |
| Sciences humaines et sociales | 12% |
| Autres sciences et techniques | 4% |
| Administration, services et tertiaires | 11% |
| Economie, commerce, industrie | 1% |
| Médias et communication | 3% |
| Art et artisanat | 1% |
| Par tranches d'âge | |
| Plus de 70 ans | 14% |
| de 50 à 70 ans | 53% |
| de 30 à 50 ans | 29% |
| moins de 30 ans | 4% |
| Par motivation | |
| Patients | 5% |
| Proche ou association de patients | 3% |
| Thèse ou études en cours | 4% |
| Intérêt professionnel | 65% |
| Simple curiosité | 23% |
Si vous n'êtes pas abonné
C'est ici
INUTILE si vous vous êtes déjà abonné, car vous le restez tant que vous ne demandez pas votre désisncription
Médecine évolutionniste (ou darwinienne)
Depuis quelques années, le problème de l'antibiorésistance, les progrès de la génomique, la redécouverte du microbiote et la prise en charge de maladies au long cours, nécessitent l'introduction d'une pensée évolutionniste dans la réflexion clinique.
Le premier diplôme universitaire intitulé "Biologie de l'évolution et médecine" a été mis en place à la faculté de Lyon en 2016.
RARE
Site médical sans publicité
et sans conflit d'intérêts.
Livres de biologie et médecine évolutionnistes
Le temps des pères - Sarah Blaffer Hrdy ▪ La Découverte, août 2025 Sarah Hrdy est connue pour avoir démontré [...]
Du bon sens dans notre assiette - Anthony Berthou ▪ Actes Sud, février 2023 Les ouvrages sur la nutrition abondent, et [...]
Comment nous sommes devenus ce que nous sommes - David Reich ▪ Quanto, 2019 La science de l'ADN ancien, née dans les années 2000, a [...]
Destinées improbables
Le hasard, la nécessité et l'avenir de l'évolution - Jonathan B Losos ▪ La Découverte, 2021
Ce livre aborde trois aspects de la biologie de [...]
L'origine des troubles mentaux - Randolph Nesse ▪ Markus Haller, 2021 Randolph Nesse et George Williams ont été les premiers [...]
Vous aimerez aussi ces humeurs...
Du protège-lame au gilet jaune - De nombreux accidents domestiques et de travail ont jalonné l’ère industrielle. L’alcool [...]
Dans l’imbroglio des comorbidités - Le 21 mars 2025, les médias, alertés par la préfecture de la Réunion et l’AFP, [...]
Grand pas dans l’histoire des médicaments - Lorsque l’on ignorait la cause des maladies, on essayait d’en soigner les symptômes. La [...]
Personnalisation sans individu - Toute décision thérapeutique s’appuie sur trois composantes. La première est évidemment [...]
Essai sur les électrohypersensibles - Les téléphones portables et leurs inélégantes antennes-relais étaient déjà la proie [...]
Vous aimerez aussi...
Naissance de la vie complexe en laboratoire - Des scientifiques recréent la danse microbienne qui a donné naissance à la vie [...]
Longévité des mammifères : gros cerveau et gènes de l’immunité - Pourquoi les chats vivent-ils généralement plus longtemps que les chiens ? De nouvelles [...]
Auto-immunité et additifs alimentaires - L'incidence des maladies auto-immunes augmente avec l'expansion de la transformation industrielle [...]
Relation entre créativité et troubles de l'humeur - Résumé La recherche conçue pour examiner la relation entre la créativité et les maladies [...]
Ocytocine synthétique et dépression du post-partum - Hypothèse En raison de ses effets puissants sur le comportement social, y compris le comportement [...]
La phrase biomédicale aléatoire
L'analyse mathématique devient un instrument aveugle si on ne la retrempe de temps en temps au foyer de l'expérience.
― Claude Bernard
Ouvrages de Luc Perino
Sapiens chez le bobologue - Luc Perino ▪ Ed du 81, novembre 2023 C'est la quatrième édition du désormais célèbre [...]
Les Non-maladies - Luc Perino ▪ Seuil, janvier 2023 La médecine moderne a indubitablement accompli de grands [...]
Patients zéro - Histoires inversées de la médecine - Luc Perino ▪ La Découverte, mars 2020 L'Histoire célèbre les victoires que les médecins [...]
La Sagesse du Médecin - Luc Perino ▪ Editions du 81, juillet 2020 Luc Perino est un auteur déjà apprécié des [...]
Darwin et les sciences de l'évolution pour les nuls - Luc Perino ▪ Pour les nuls 2018 Voici une passionnante histoire de la vie et du genre [...]